Chapter 7 Working on remote servers
7.1 Accessing remote computers
The primary protocol for accessing remote computers in this day and age
is ssh which stands for “Secure Shell.” In the protocol, your computer
and the remote computer talk to one another and then choose to have a “shared
secret” which they can use as a key to encrypt data traffic from one to the other.
The amazing thing is that the two computers can actually tell each other what
that shared secret is by having a conversation “in the open” with one another.
That is a topic for another day, but if you are interested, you could
read about it here.
At any rate, the SSH protocal allows for secure access to a remote server. It involves using a username and a password, and, in many cases today, some form of two-factor authentication (i.e., you need to have your phone involved, too!). Different remote servers have different routines for logging in to them, and they are also all configured a little differently. The main servers we are concerned about in these teaching materials are:
- The Hummingbird cluster at UCSC, which is accessible by anyone with a UCSC blue username/password.
- The Alpine Supercomputer at CU Boulder which is accessible by all graduate students and faculty at CSU.
- The Sedna cluster housed at the National Marine Fisheries Service, Northwest Fisheries Science Center. This is accessible only by those Federal NMFS employees whom have been granted access.
Happily, all of these systems use SLURM for job scheduling (much more about that in the next chapter); however are a few vagaries to each of these systems that we will cover below.
7.1.1 Windows
If you are on a Windows machine, you can use the ssh utility from your Git Bash shell, but
that is a bit of a hassle from RStudio. And a better terminal emulator is available if you
are going to be accessing remote computers. It is recommended that you install and
use the program PuTTy. The steps are pretty self-explanatory
and well documented. Instead of using ssh on a command line you put a host name into
a dialog box, etc.
WHOA! I’m not a Windows person, but I just Matthew Hopken working on Windows using MobaXterm to connect to the server and it looks pretty nice.
7.1.2 Hummingbird
Directions for UCSC students and staff to login to Hummingbird are available at https://www.hb.ucsc.edu/getting-started/. If you are not on the UCSC campus network, you need to use the UCSC VPN to connect.
By default, this cluster uses tcsh for a shell rather than bash. To keep things
consistent with what you have learned about bash, you will want to automatically switch
to bash upon login. You can do this by adding a file ~/.tcshrc whose contents are:
Then, configure your bash environment with your ~/.bashrc and ~/.bash_profile as
described in Chapter 4.5.
The tmux settings (See section 7.4) in hummingbird are a little messed up as well, making
it hard to set window names that don’t get changed the moment you make another command. Therefore,
you must make a file called ~/.tmux.conf and put this line in it:
set-option -g allow-rename off7.1.3 Alpine
To get an account on the CU Boulder computing resources (which includes Alpine), see https://www.acns.colostate.edu/hpc/summit-get-started/. Account creation is automatic for graduate students and faculty. This setup requires that you get an app called Duo on your phone for doing two-factor authentication.
Instructions for logging into Summit are at https://www.acns.colostate.edu/hpc/#remote-login.
On your local machine (i.e., laptop), you might consider adding an alias to your
.bashrc that will let you type summit to issue the login command. For example:
where you replace csu_eid with your actual CSU eID.
7.2 Transferring files to remote computers
7.2.1 sftp and several systems that use it
Most Unix systems have a command called scp, which works like cp, but which is
designed for copying files to and from
remote servers using the SSH protocol for security. This works really well
if you have set up a public/private key pair to allow SSH access to your server
without constantly having to type in your password. Use of public-private keypairs is unfortunately, not
an option (as far as I can tell) on new NSF-funded clusters that use 2-factor authentication (like SUMMIT
at CU Boulder). Trying to use scp in such a context becomes an endless cycle of
entering your password and checking your phone for a DUO push. Fortunately, there are
alternatives.
7.2.1.1 Vanilla sftp
The administrators of the SUMMIT supercomputer at CU Boulder recommend
the sftp utility for transferring files from your laptop to the server.
This works reasonably well. The syntax for a CSU student or affiliate connecting to the server is
sftp csu_userEID@colostate.edu@login.rc.colorado.edu
# for example, here is mine:
sftp eriq@colostate.edu@login.rc.colorado.eduAfter doing this you have to give your eID password followed by ,push, and then
approve the DUO push request on your phone. Once that is done, you have a “line open”
to the server and can use the commands of sftp to transfer files around.
However, the vanilla version of sftp (at least on a Mac) is unbelievably limited,
because there is simply no good support for TAB completion within
the utility for navigating directories on the server or upon your laptop.
It must have been developed by troglodytes…consequently, I won’t describe
vanilla sftp further.
7.2.1.2 Windows alternatives
If you are on Windows, it looks like the makers of PuTTY also bring you PSFTP which might be useful for you for file transfer. Even better, MobaXterm has native GUI file transfer capabilities. Go for it!
7.2.1.3 A GUI solution for Mac or Windows
When you are first getting started transfering files to a server, it might be easiest to use a graphical user interface. There is a decently-supported (and freely available) application called FileZilla, that does this. You can download the FileZilla client application appropriate for your operating system (note! you download and install this on your own laptop not the server) from https://filezilla-project.org/download.php?type=client.
Once you install it, there are a few configurations to be done. First, go toEdit->Settings and activate
and give a master password to protect your passwords. This master password should be something that
you will remember easily. It does not have to be, and, really, should not be, the same as your Summit password.
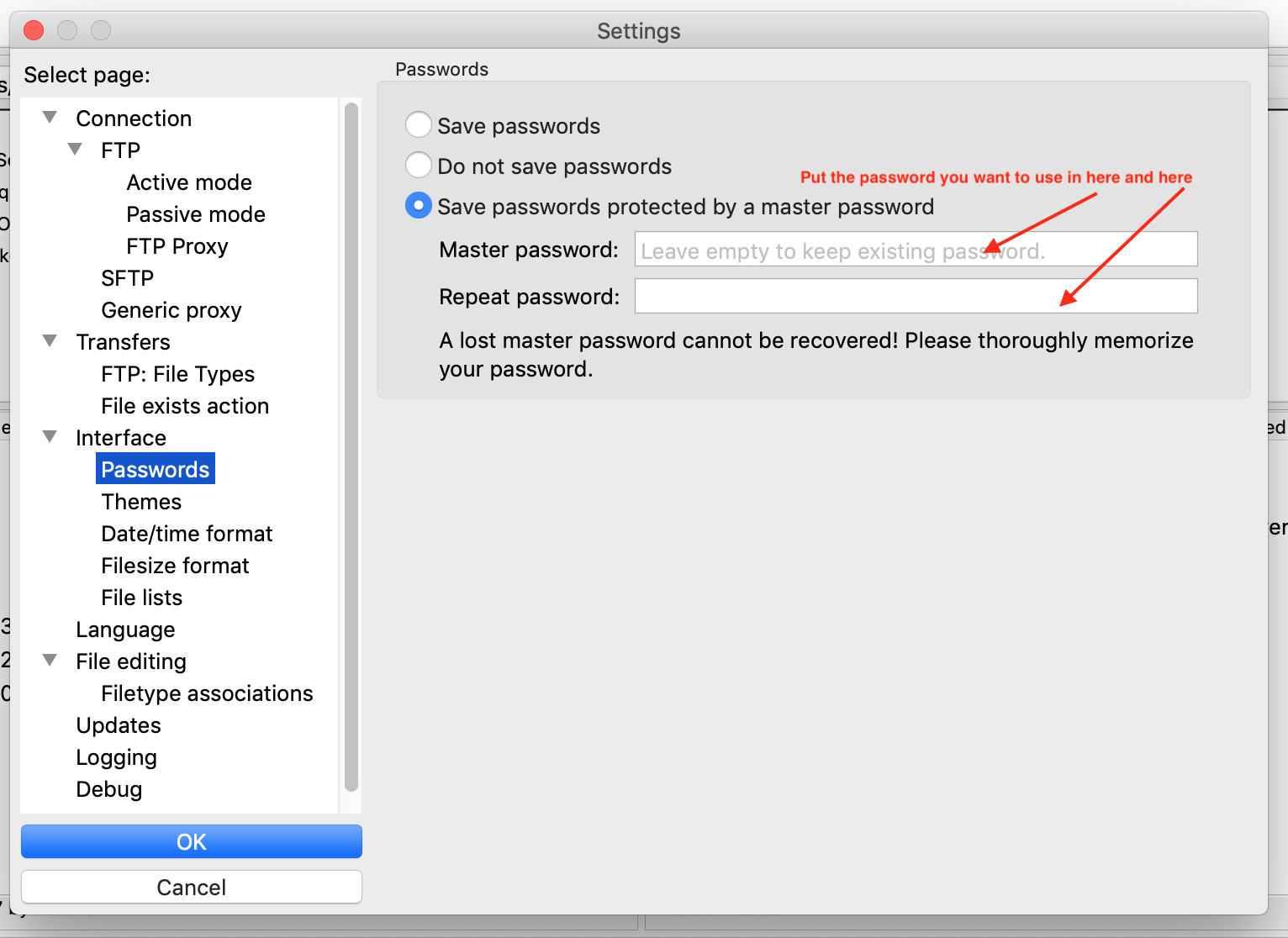
FIGURE 7.1: Setting FileZilla’s master password
Edit->Settings request
FIGURE 7.2: Setting FileZilla’s master password
File->Site Manager and set up a connection to your remote machine.
For SUMMIT, do like this:
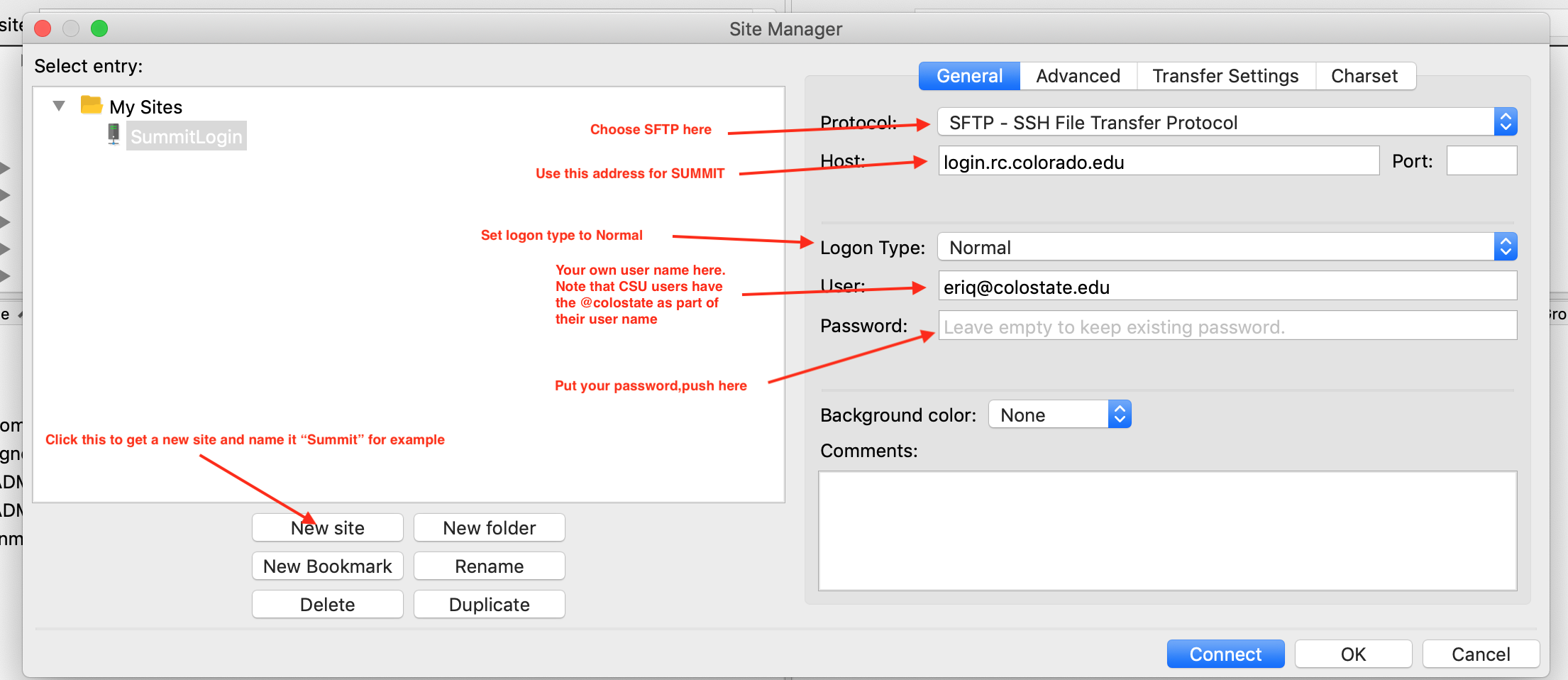
FIGURE 7.3: Setting FileZilla’s master password
After you hit OK and have established this site, you can do File->Site Manager, then choose
your Summit connection in the left pane and hit “connect” to connect to Summit. You may have to
type in the “Master Password” that you gave to FileZilla.
After connecting, you have two file-browser panes. The one on your left is typically your local computer, and the one on the right is the server (remote computer). You can change the local or remote directory by clicking in either the left or right pane, and files and folders by dragging and dropping. The setup looks like this:
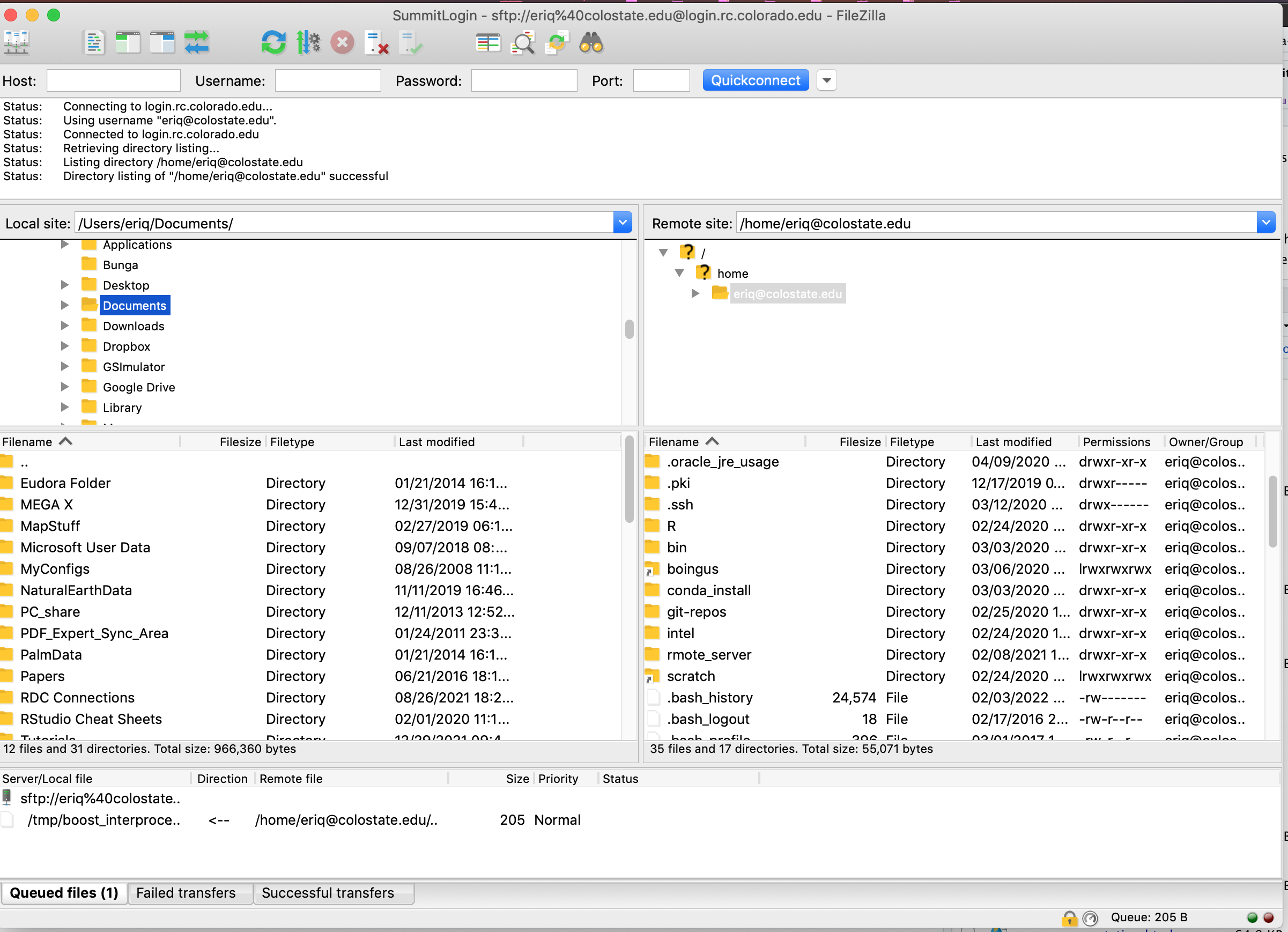
FIGURE 7.4: Setting FileZilla’s master password
7.2.1.4 lftp
If you are on a Mac, you can install lftp (brew install lftp: note that I need to write
a section about installing command line utilities via homebrew somewhere in this handbook).
lftp provides the sort of TAB completion of paths that you, by now, will have come to
know and love and expect.
Before you connect to your server with lftp there are a few customizations that you will
want to do in order to get nicely colored output, and to avoid having to login repeatedly
during your lftp session. You must make a file on your laptop called ~/.lftprc and put
the following lines in it:
set color:dir-colors "rs=0:di=01;36:fi=01;32:ln=01;31:*.txt=01;35:*.html=00;35:"
set color:use-color true
set net:idle 5h
set net:timeout 5hNow, to connect to SUMMIT with lftp, you use this syntax (shown for my username):
That can be a lot to type, so I would recommend putting something this in your
.bashrc:
so you can just type summit_ftp (which will TAB complete…) to launch that command.
After you issue that command, you put in your password (on SUMMIT, followed by ,push). lftp then caches your
password, and will re-issue it, if necessary, to execute commands. It doesn’t actually send your
password until you try a command like cls. On the SUMMIT system, with the default lftp settings,
after 3 minutes of idle time, when you issue an sftp command on the server, you will have to approve
access with the DUO app on your phone again. However, the line last two lines in the
~/.lftprc file listed above ensure that your connection to SUMMIT will stay active even
through 5 hours of idle time, so you don’t have to keep clicking DUO pushes on your phone.
After 5 hours, if you try issuing a command to the server in lftp, it will use your cached
password to reconnect to the server. On SUMMIT, this means that you only need to deal with
approving a DUO push again—not re-entering your password. If you are working on SUMMIT daily,
it makes sense to just keep one Terminal window open, running lftp, all the time.
Once you have started your lftp/sftp session this way, there are some important things to keep in mind.
The most important of which is that the lftp session you are in maintains a current working directory
on both the server and on your laptop. We will call these the server working directory and
the laptop working directory, respectively, (Technically, we ought to call the laptop working directory the client working directory
but I find that is confusing for people, we we will stick with laptop.)
There are two different commands to see what each
current working directory is:
pwd: print the server working directorylpwd: print laptop working directory (the precedinglstands for local).
If you want to change either the server or the laptop current working directory you use:
cdpath : change the server working directory to pathlcdpath : change the laptop working directory to path.
Following lcd, TAB-completion is done for paths on the laptop, while following
cd, TAB-completion is done for paths on the server.
If you want to list the contents of the different directories on the servers you use:
cls: list things in the server working directory, orclspath : list things in path on the server.
Note that cls is a little different than the ls command that comes
with sftp. The latter command always prints in long format and does not play
nicely with colorized output. By contrast, cls is part of lftp and it
behaves mostly like your typical Unix ls command, taking options like -a, -l and -d, and
it will even do cls -lrt. Type help cls at the lftp prompt for more information.
If you want to list the contents of the different directories on your laptop, you
use ls but you preface it with a !, which means “execute the following on my
laptop, not the server.” So, we have:
!ls: list the contents of the laptop working directory.!lspath : list the contents of the laptop path path.
When you use the ! at the beginning of the line, then all the TAB completion occurs
in the context of the laptop current working directory. Note that with the !
you can do all sorts of typical shell commands on your laptop from within the lftp
session. For example !mkdir this_on_my_laptop or !cat that_file, etc.
If you wish to make a directory on the server, just use mkdir. If you wish to
remove a file from the server, just use rm. The latter works much like it does in
bash, but does not seem to support globbing (use mrm for that!) In fact, you can
do a lot of things (like cat and less) on the server
as if you had a bash shell running on it through an
SSH connection. Just type those commands at the lftp prompt.
7.2.1.5 Transferring files using lftp
To this point, we haven’t even talked about our original goal with lftp, which
was to transfer files from our laptop to the server or from the server to our laptop.
The main lftp commands for those tasks are: get, put, mget, mput, and mirror—it is
not too much to have to remember.
As the name suggests, put is for putting files from your laptop onto the server. By default it
puts files into the server working directory. Here is an example:
If you want to put the file into a different directory on the server (that must already exist)
you can use the -O option:
The command get works in much the same way, but in reverse: you are getting things
from the server to your laptop. For example:
# copy to laptop working directory
get serverFile_1 serverFile1_2
# copy to existing directory laptop_dest_dir
get -O laptop_dest_dir serverFile_1 serverFile1_2Neither of the commands get or put do any of the pathname expansion (or “globbing” as it
we have called it) that you will be familiar with from the bash shell. To effect that sort
of functionality you must use mput and mget, which, as the m prefix in the
command names suggests, are the “multi-file” versions of put and get. Both of these
commands also take the -O option, if desired, so that the above commands could be
rewritten like this:
Finally, there is not a recursive option, like there is with cp, to any of get, put, mget,
or mput. Thus, you cannot use any of those four to put/get entire directories on/from the
server. For that purpose, lftp has reserved the mirror command. It does what it sounds like:
it mirrors a directory from the server to the laptop. The mirror command can actually
be used in a lot of different configurations (between two remote servers, for example) and
with different settings (for example to change only pre-existing files older than
a certain date).
However, here, we will demonstrate only its common use case
of copying directories between a server and laptop here.
To copy a directory dir, and its contents, from your server to your
laptop current directory you use:
To copy a directory ldir from your laptop to your server current directory you
use -R which transmits the directory in the reverse direction:
Learning to use lftp will require a little bit more of your time, but it is worth
it, allowing you to keep a dedicated terminal window open for file transfers with sensible
TAB-completion capability.
7.2.2 git
Most remote servers you work on will have git by default.
If you are doing all your work on a project within a single
repository, you can use git to keep scripts and other files
version-controlled on the server. You can also push and pull files
(not big data or output files!) to GitHub, thus keeping things backed up
and version controlled, and providing a useful way to synchronize scripts
and other files in your project between the server and your laptop.
Example:
- write and test scripts on your laptop in a repo called
my-project - commit scripts on your laptop and push them to GitHub in a repo also
called
my-project - pull
my-projectfrom GitHub to the server. - Try running your scripts in
my-projecton your server. In the process, you may discover that you need to change/fix some things so they will run correctly on the server. Fix them! - Once things are fixed and successfully running on the server, commit those changes and push them to GitHub.
- Update the files on your laptop so that they reflect the changes you
had to make on the server, by pulling
my-projectfrom GitHub to your laptop.
7.2.2.1 Configuring git on the remote server
In order to make this sort of worklow successful, you first need to ensure that you have set up git on your remote server. Doing so involves:
- establishing your name and email that will be used with your git commits made from the server.
- Ensuring that git password caching is set up so you don’t always have to type your GitHub password when you push and pull.
- configuring your git text editor to be something that you know how to use.
It can be useful give yourself a git name on the server that reflects the fact that the changes you are committing were made on the server.
For example, for my own setup on the Summit cluster at Boulder, I might do my git configurations by issuing these commands on the command line on the server:
git config --global user.name "Eric C. Anderson (From Summit)"
git config --global user.email eriq@rams.colostate.edu
git config --global core.editor nanoIn all actuality, I tend to set my editor to be vim or emacs, because those are
more powerful editors and I am familiar with then; however, if you are new to Unix,
then nano is an easy-to-use editor, and one is less likely to get “stuck” inside of it, as can happen in vim.
You should set configurations on your server appropriate to yourself (i.e., with your name and email and preferred text editor). Once these configurations are set, you are ready to start cloning repositories from GitHub and then pushing and pulling them, as well.
To this point, we have always done those actions from within RStudio. On a remote server, however, you will have to do all these actions from the command line. That is OK, it just requires learning a few new things.
The first, and most important, issue to understand is that if you want to push new changes back to a repository that is on your GitHub account, GitHub needs to know that you have privileges to do so. Back in the days when you could make authenticated https connections to GitHub, there were some tricks to this. But, since all your connections to GitHub must now be done with SSH, it has actually gotten a lot easier (but it involves setting up SSH keys, as described in the next section).
7.2.2.2 Using git on the remote server
When on the server, you don’t have the convenient RStudio interface to git, so you have to use git commands on the command line. Fortunately these provide straightforward, command-line analogies to the RStudio GUI git interface you have become familiar with.
Intead of having an RStudio Git panel that shows you files that are new or
have been modified, etc., you use git status in your repo to give
a text report of the same.
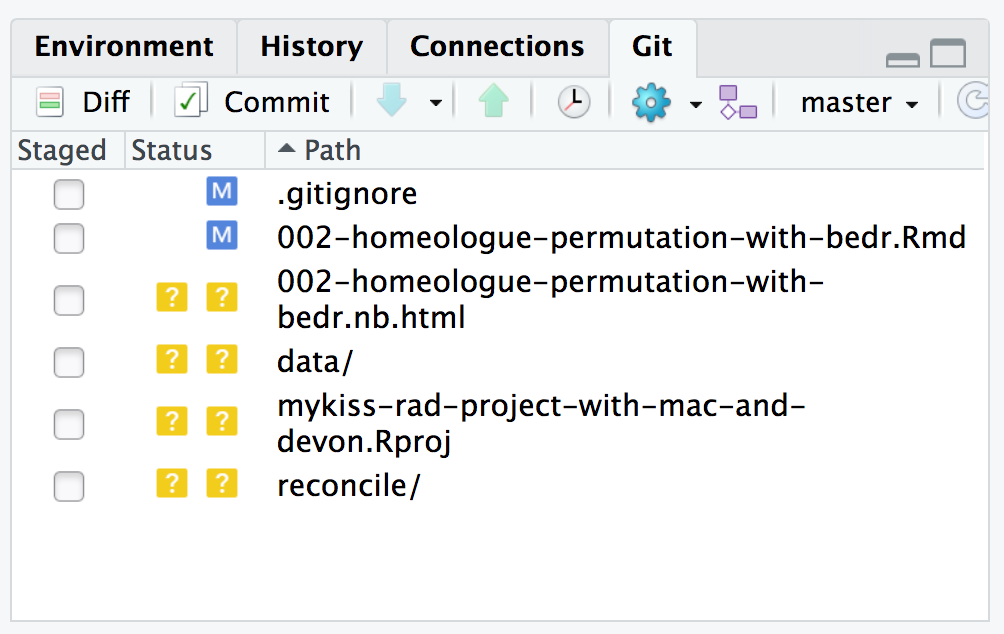
FIGURE 7.5: An example of what an RStudio git window might look like.
That view is merely showing you a graphical view of the output of
the git status command run at the top level of the repository which
looks like this:
% git status
On branch master
Changes not staged for commit:
(use "git add <file>..." to update what will be committed)
(use "git checkout -- <file>..." to discard changes in working directory)
modified: .gitignore
modified: 002-homeologue-permutation-with-bedr.Rmd
Untracked files:
(use "git add <file>..." to include in what will be committed)
002-homeologue-permutation-with-bedr.nb.html
data/
mykiss-rad-project-with-mac-and-devon.Rproj
reconcile/
no changes added to commit (use "git add" and/or "git commit -a")Aha! Be sure to read that and understand that the output tells you which files are tracked by git and Modified (blue M in RStudio) and which are untracked (Yellow ? in RStudio).
If you wanted to see a report of the changes in the files relative
to the currently committed version, you could use git diff, passing
it the file name as an argument. We will see an example of that below…
git you first must
stage them. In RStudio you do that by clicking the little button to
the left of the file or directory in the Git window. For example,
if we clicked the buttons for the data/ directory, as well as for
.gitignore and 002-homeologue-permutation-with-bedr.Rmd, we would
have staged them and it would look like Figure 7.6.
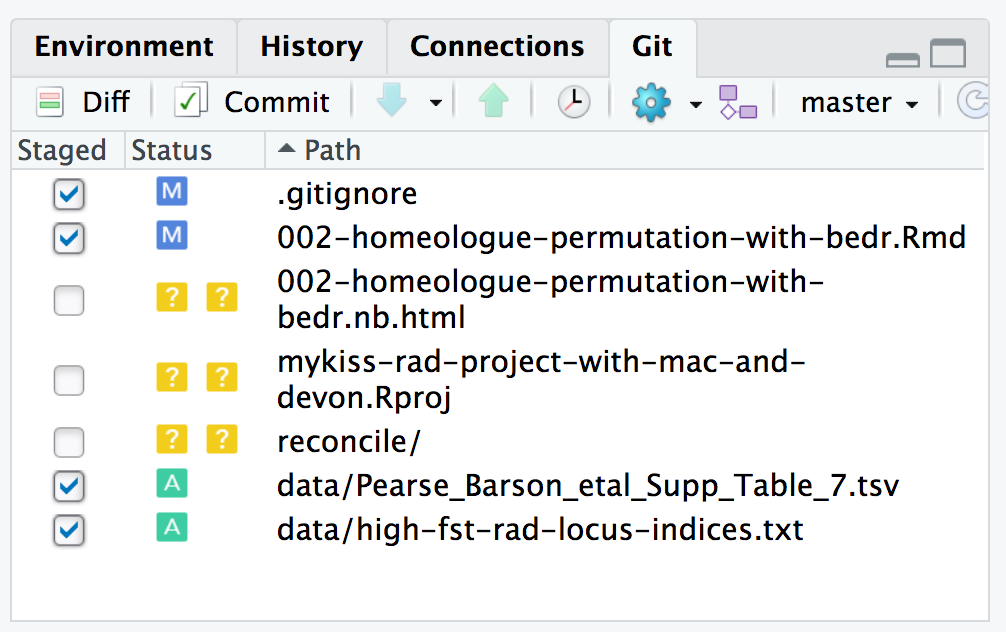
FIGURE 7.6: An example of what an RStudio git window might look like.
In order to do the equivalent operations with git on the command line
you would use the git add command, explicitly naming the files you wish to
stage for committing:
Now, if you check git status you will see:
% git status
On branch master
Changes to be committed:
(use "git reset HEAD <file>..." to unstage)
modified: .gitignore
modified: 002-homeologue-permutation-with-bedr.Rmd
new file: data/Pearse_Barson_etal_Supp_Table_7.tsv
new file: data/high-fst-rad-locus-indices.txt
Untracked files:
(use "git add <file>..." to include in what will be committed)
002-homeologue-permutation-with-bedr.nb.html
mykiss-rad-project-with-mac-and-devon.Rproj
reconcile/It tells you which files are ready to be committed!
In order to commit the files to git you do:
And then, to push them back to GitHub (if you cloned this repository from GitHub), you can simply do:
That syntax is telling git to push the master branch (which is
the default branch in a git repository), to the repository labeled as
origin, which will be the GitHub repository if you cloned the repository
from GitHub. (If you are working with a different git branch than master,
you would need to specify its name here. That is not difficult, but is
beyond the scope of this chapter.)
Now, assuming that we cloned the alignment-play repository to our
server, here are the steps involved in editing a file, committing the
changes, and then pushing them back to GitHub. The command in the following
is written as [alignment-play]--% which is telling us that we are in the
alignment-play repository.
# check git status
[alignment-play]--% git status
# On branch master
nothing to commit, working directory clean
# Aha! That says nothing has been modified.
# But, now we edit the file alignment-play.Rmd
[alignment-play]--% nano alignment-play.Rmd
# In this case I merely added a line to the YAML header.
# Now, check status of the files:
[alignment-play]--% git status
# On branch master
# Changes not staged for commit:
# (use "git add <file>..." to update what will be committed)
# (use "git checkout -- <file>..." to discard changes in working directory)
#
# modified: alignment-play.Rmd
#
no changes added to commit (use "git add" and/or "git commit -a")
# We see that the file has been modified.
# Now we can use git diff to see what the changes were
[alignment-play]--% git diff alignment-play.Rmd
diff --git a/alignment-play.Rmd b/alignment-play.Rmd
index 9f75ebb..b389fae 100644
--- a/alignment-play.Rmd
+++ b/alignment-play.Rmd
@@ -3,6 +3,7 @@ title: "Alignment Play!"
output:
html_notebook:
toc: true
+ toc_float: true
---
# The output above is a little hard to parse, but it shows
# the line that has been added: " toc_float: true" with a
# "+" sign.
# In order to commit the changes, we do:
[alignment-play]--% git add alignment-play.Rmd
[alignment-play]--% git commit
# after that, we are bumped into the nano text editor
# to write a short message about the commit. After exiting
# from the editor, it tells us:
[master 001e650] yaml change
1 file changed, 1 insertion(+)
# Now, to send that new commit to GitHub, we use git push origin master
[alignment-play]--% git push origin master
Password for 'https://eriqande@github.com':
Counting objects: 5, done.
Delta compression using up to 24 threads.
Compressing objects: 100% (3/3), done.
Writing objects: 100% (3/3), 325 bytes | 0 bytes/s, done.
Total 3 (delta 2), reused 0 (delta 0)
remote: Resolving deltas: 100% (2/2), completed with 2 local objects.
To https://eriqande@github.com/eriqande/alignment-play
0c1707f..001e650 master -> masterIn order to push to a GitHub repository from your remote server you will
need to establish a public/private SSH key pair, and share the public key
in the settings of your GitHub account. The process for this is similar to
what you have already done for accessing GitHub via git with your laptop:
follow the directions for Linux systems at:
https://happygitwithr.com/ssh-keys.html.
In order to copy your public key to GitHub, it will be easiest to
cat ~/.ssh/id_ed25519.pub to stdout and then copy it from your terminal to
GitHub.
Finally, if after pushing those changes to GitHub, we then pull them
down to our laptop, and make more changes on top of them and push those
back to GitHub, we can retrieve from GitHub to the server those changes we
made on our laptop with git pull origin master. In other words, from the
server we simply issue the command:
7.2.3 Globus
Globus is a file transfer system for high performance computing that was developed long ago by a group at the University of Chicago. If you work at an institution that has a subscription to the Globus system (as is the case with Colorado State University!), then it is quite easy to use it.
In the Globus model, files get transferred between different “endpoints.” Which are typically file servers on large university computing systems. You as a user are entitled to initiate transfers between the endpoints to which you have access rights. You can initiate these transfers using a web interface through your web browser. This makes is incredibly convenient, especially if you want to transfer large files between different computing clusters that are endpoints on the Globus network.
Addtionally, Globus provides a small software application that can turn your own laptop or your desktop workstation into a Globus endpoint, allowing you to initiate data transfers between your laptop/desktop and the cluster. Globus is a well-tested and robust system, so, since it is offered for Colorado State University students and faculty, it is well worth using.
The steps to using it are:
Sign in to Globus as a Colorado State Affiliate by going to https://www.globus.org/app/login, and finding Colorado State University in the dropdown menu, and hitting continue.
 When you do that the first time, you might need to agree to using CILogin. Do so.
When you do that the first time, you might need to agree to using CILogin. Do so.You are then taken to a page to authenticate with CSU—it is the familiar eID login. Login to it. For me it looks like this:

After authenticating, you might be taken to a page that looks like this:
 To be honest, I don’t know what this is about. I think it is Globus pitching its paid
options. Whatever….You don’t need it.
To be honest, I don’t know what this is about. I think it is Globus pitching its paid
options. Whatever….You don’t need it.
Instead, proceed directly to https://app.globus.org/file-manager which looks like this:
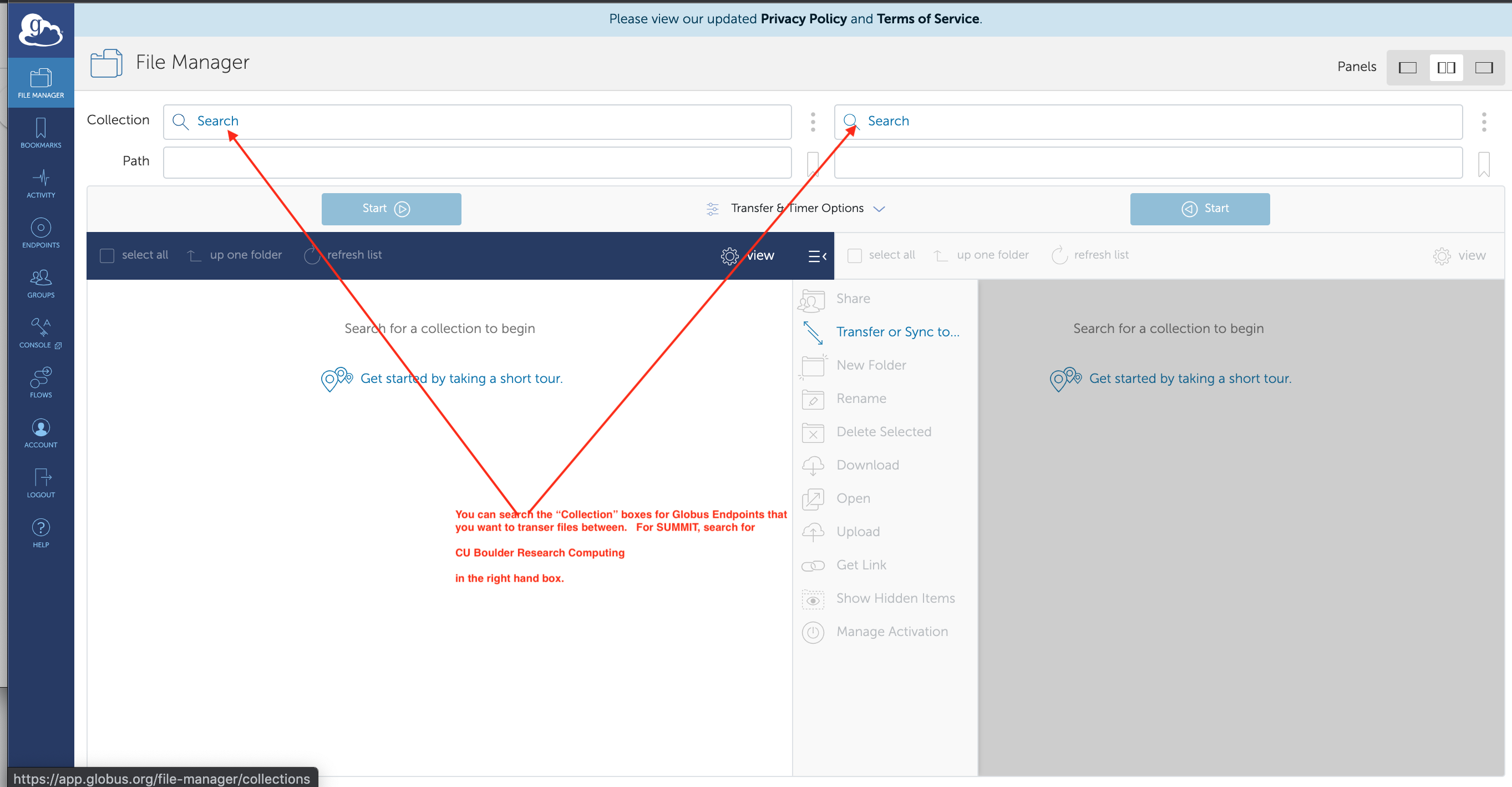 Search for
Search for CU Boulder Research Computingin the right hand box. When you find it and select it, you should see your home directory on SUMMIT in it, like this: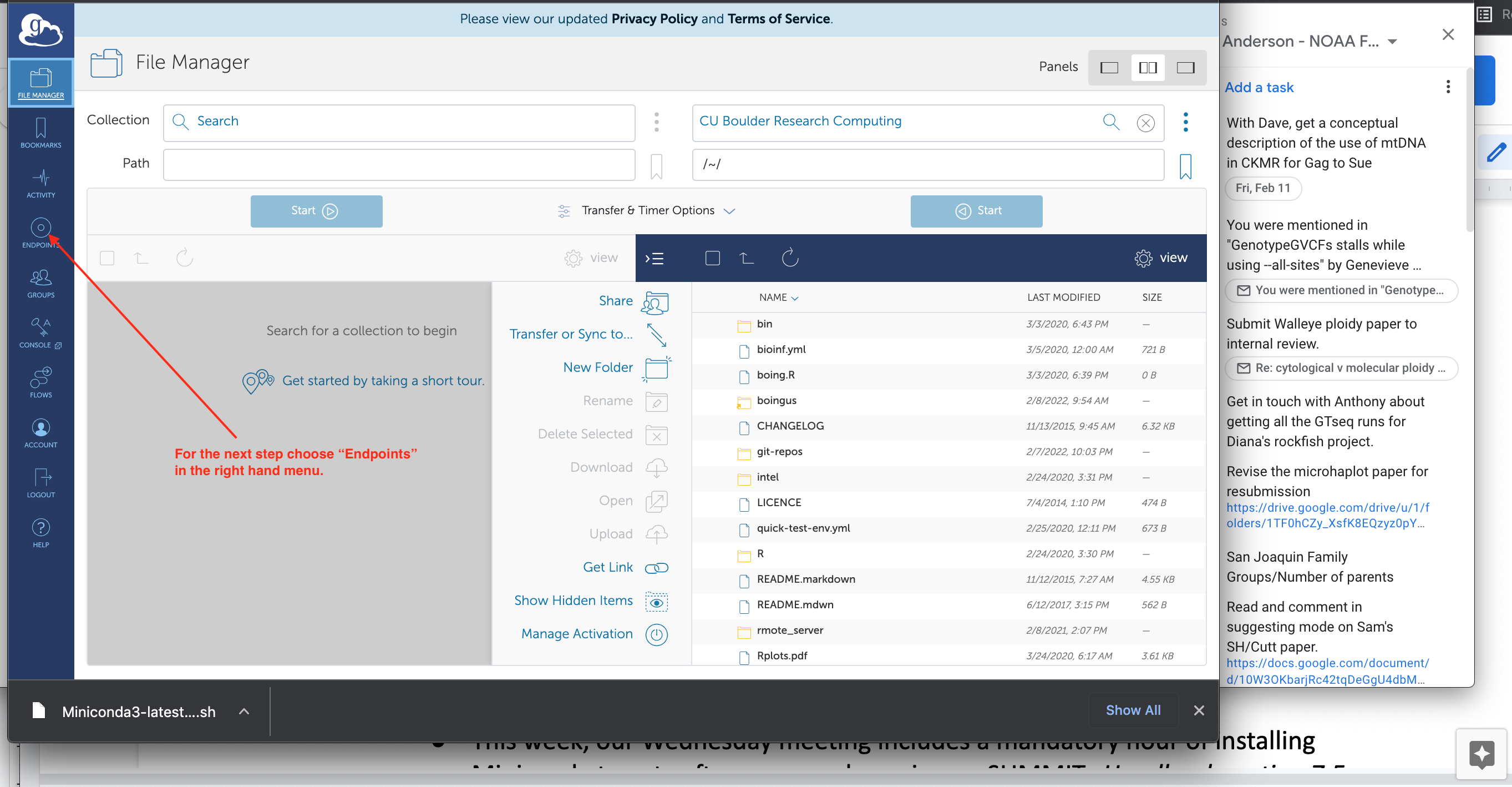
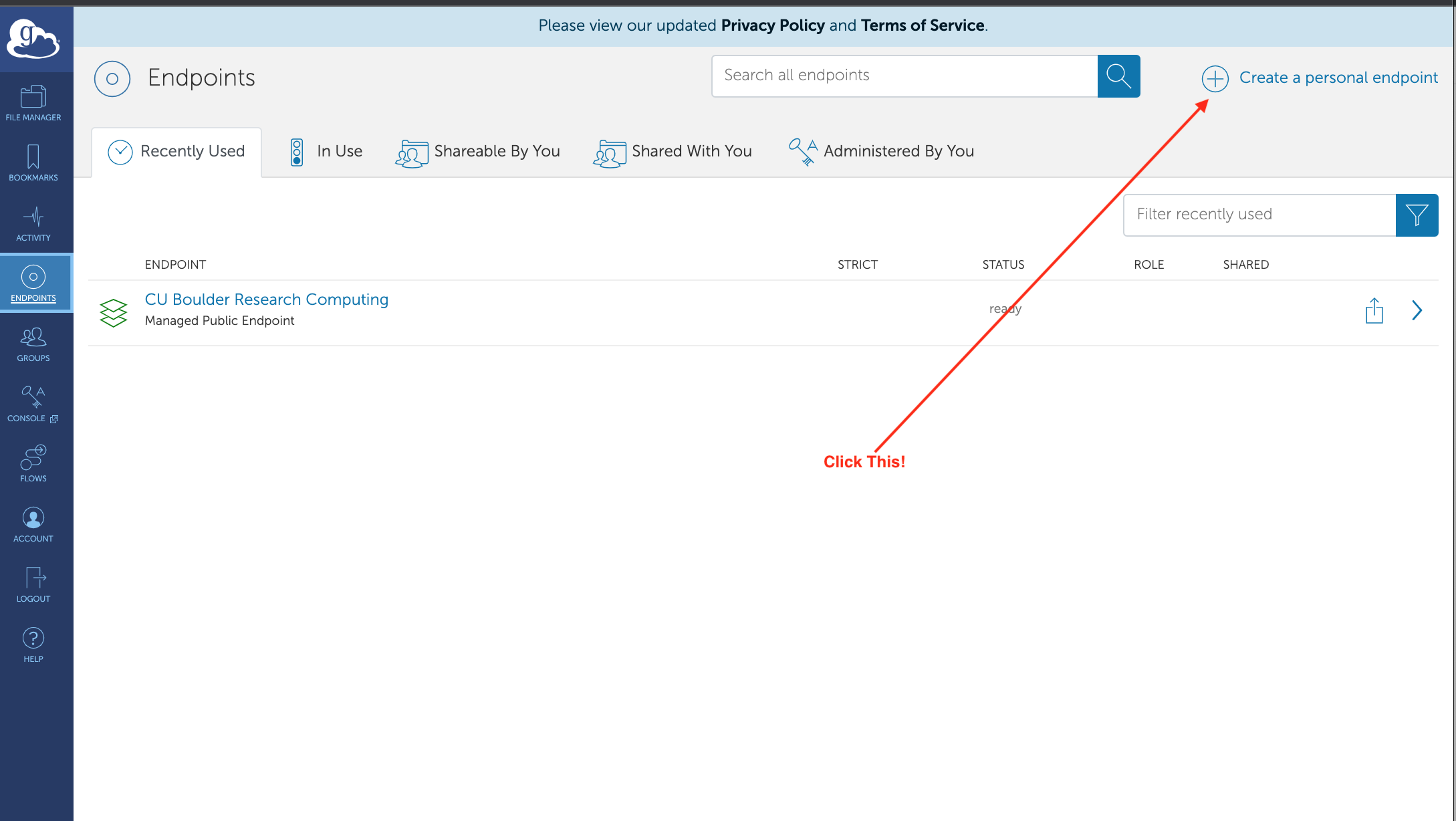
For the next step, you want to create an endpoint on your own laptop. Choose the “Endpoints” in the left menu (see red arrow in picture above). When you do that you can find the “Create a personal endpoint” link:
 After clicking that, click the link to download “Globus Personal Connect” for your
operating system.
After clicking that, click the link to download “Globus Personal Connect” for your
operating system.After downloading it, install “Globus Personal Connect”.
After installing it, open “Globus Personal Connect”. If you haven’t used it before, it should ask you to log in:
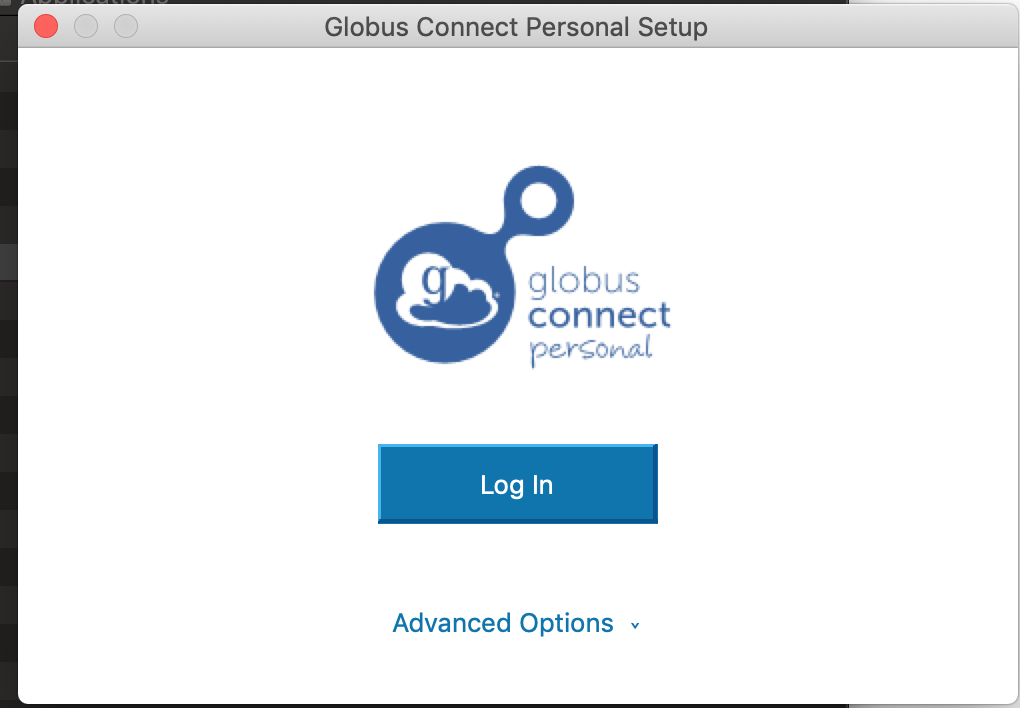
After clicking log-in enter a name by which you would like to call your endpoint, and then choose “Allow”
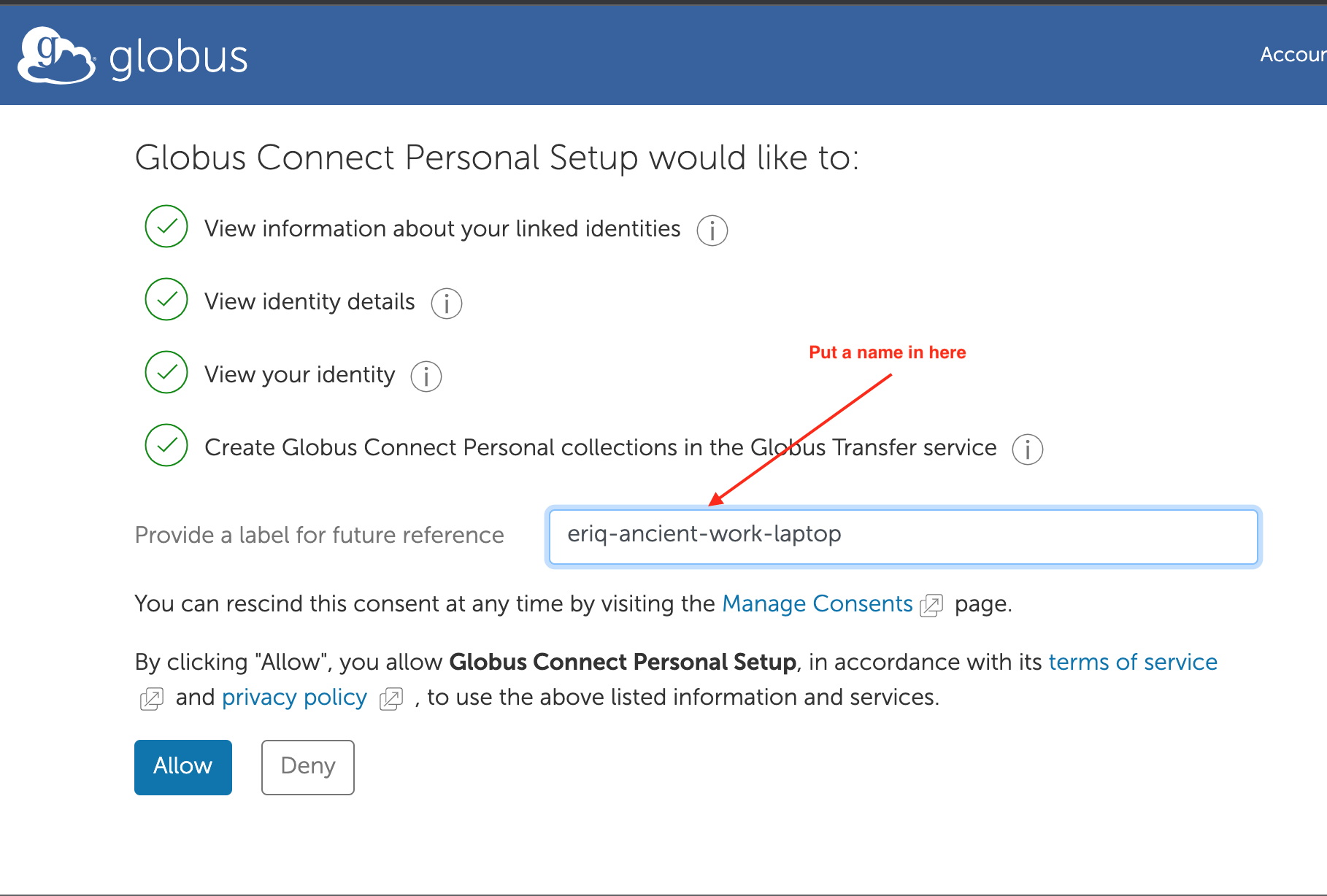
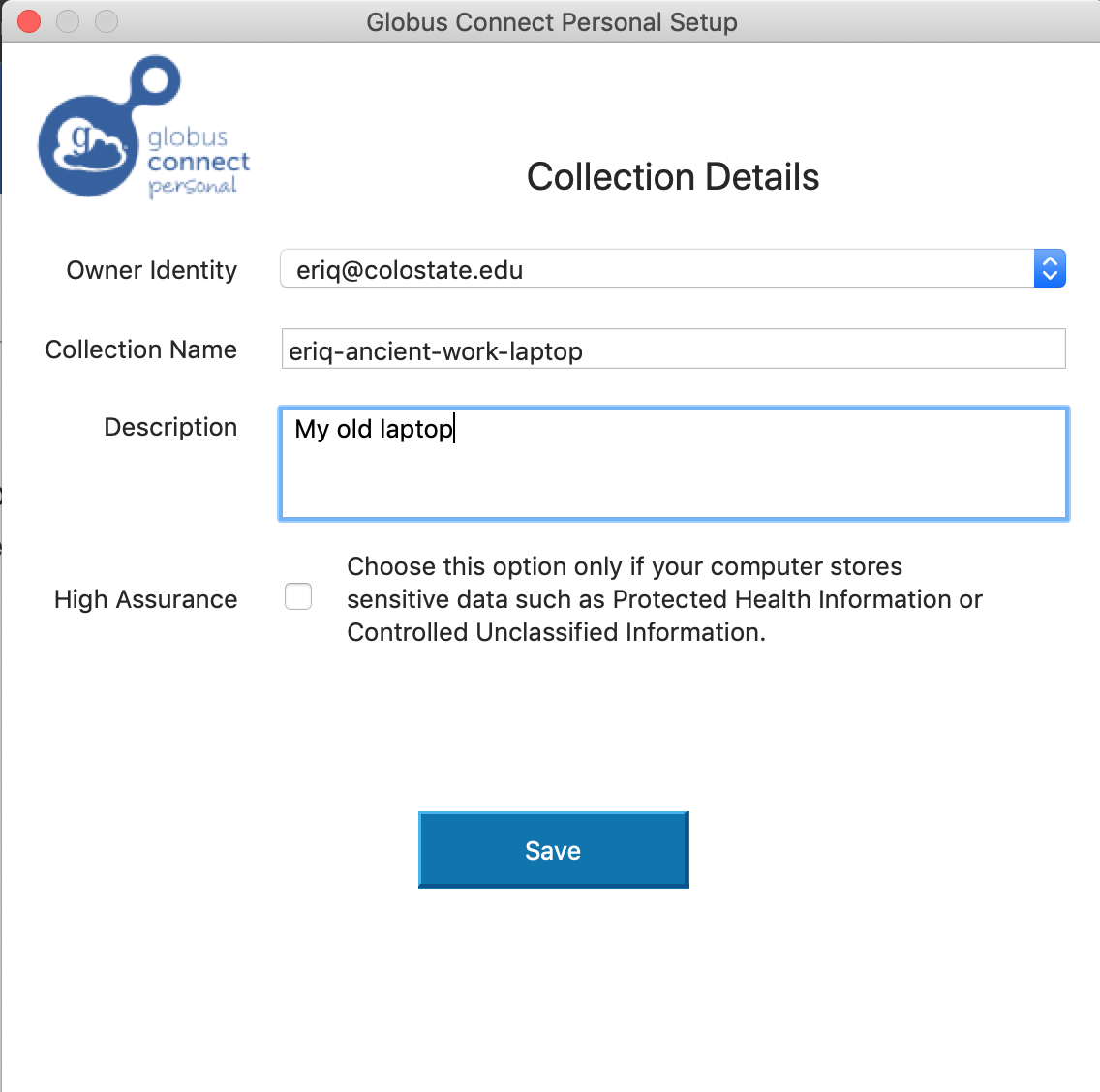
Only one more screen to go. Fill in some more names that are appropriate to your laptop/desktop and choose “Save”. (Don’t put in the names I have used…) You probably do not want to choose the High Assurance option, as that requires an extra round of work for the sys admins…
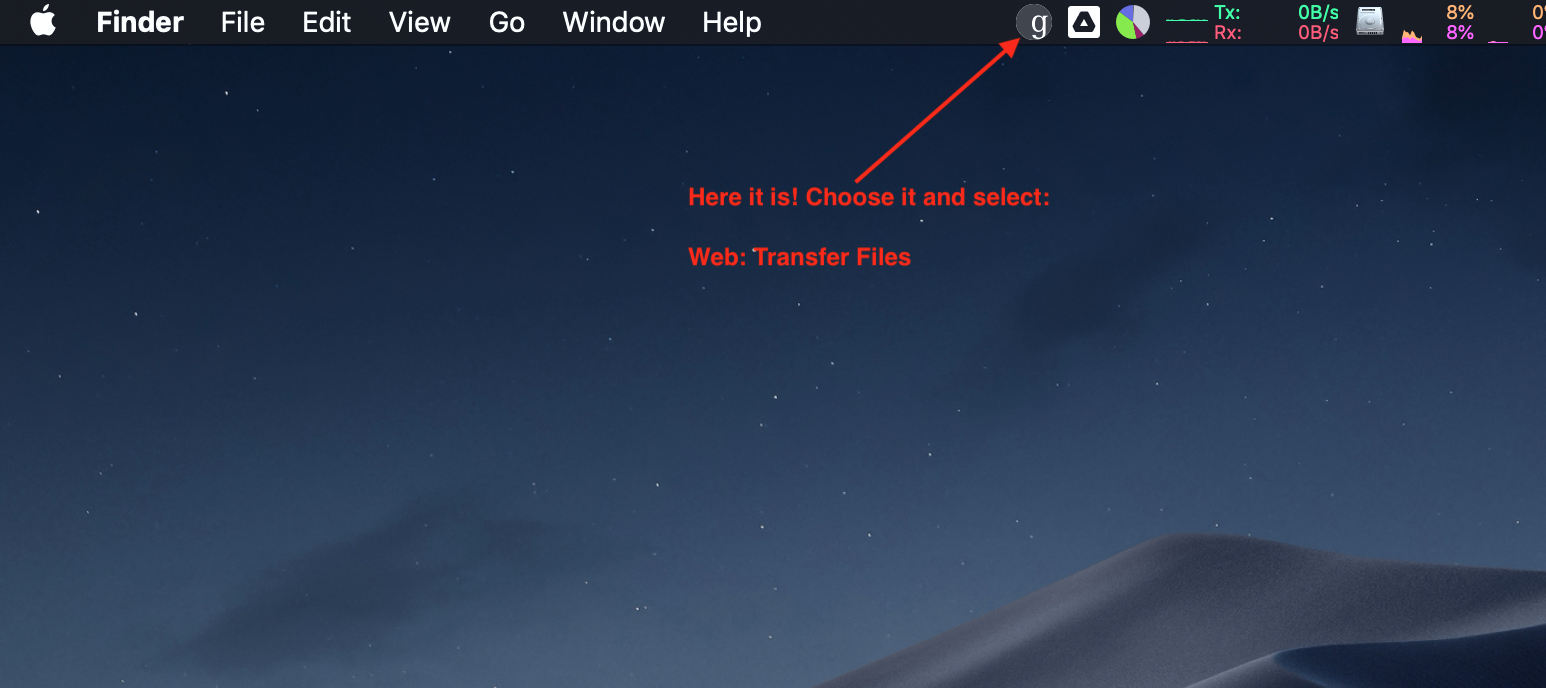
Yay! You are done. Now, on a mac, you can find the Globus icon in the menu bar and use that to start a web transfer session:
And when you get that web page, your laptop will be the left endpoint and you can search for “CU Boulder Research Computing” in the right endpoint box.
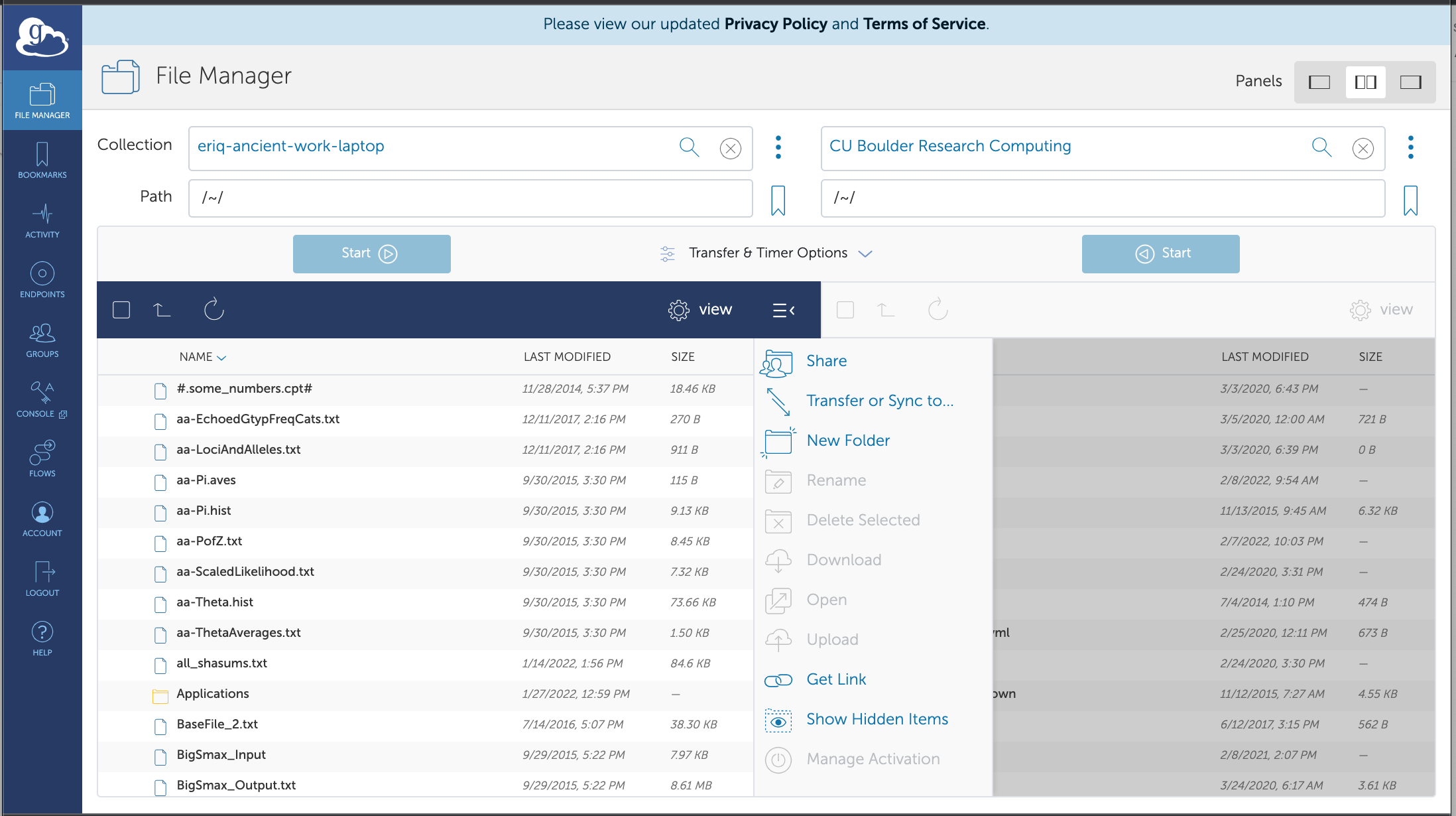
Now copying things from one to another is as easy as highlighting files from your desired source endpoint (left or right) and then hitting the “Start” button for that source endpoint.
7.2.4 Interfacing with “The Cloud”
Increasingly, data scientists and tech companies alike are keeping their data “in the cloud.” This means that they pay a large tech firm like Amazon, Dropbox, or Google to store their data for them in a place that can be accessed via the internet. There are many advantages to this model. For one thing, the company that serves the data often will create multiple copies of the data for backup and redundancy: a fire in a single data center is not a calamity because the data are also stored elsewhere, and can often be accessed seamlessly from those other locations with no apparent disruption of service. For another, companies that are in the business of storing and serving data to multiple clients have data centers that are well-networked, so that getting data onto and off of their storage systems can be done very quickly over the internet by an end-user with a good internet connection.
Five years ago, the idea of storing next generation sequencing data in the cloud might have sounded a little crazy—it always seemed a laborious task getting the data off of the remote server at the sequencing center, so why not just keep the data in-house once you have it? To be sure, keeping a copy of your data in-house still can make sense for long-term data archiving needs, but, today, cloud storage for your sequencing data can make a lot of sense. A few reasons are:
- Transferring your data from the cloud to the remote HPC system that you use to process the data can be very fast.
- As above, your data can be redundantly backed up.
- If your institution (university, agency, etc.) has an agreement with a cloud storage service that provides you with unlimited storage and free network access, then storing your sequencing data in the cloud will cost considerably less than buying a dedicated large system of hard drives for data backup. (One must wonder if service agreements might not be at risk of renegotiation if many researchers start using their unlimited institutional cloud storage space to store and/or archive their next generation sequencing data sets. My own agency’s contract with Google runs through 2021…but I have to think that these services are making plenty of money, even if a handful of researchers store big sequence data in the cloud. Nonetheless, you should be careful not to put multiple copies of data sets, or intermediate files that are easily regenerated, up in the cloud.)
- If you are a PI with many lab members wishing to access the same data set, or even if you are just a regular Joe/Joanna researcher but you wish to share your data, it is possible to effect that using your cloud service’s sharing settings. We will discuss how to do this with Google Drive.
There are clearly advantages to using the cloud, but one small hurdle remains. Most
of the time, working in an HPC environment, we are using Unix, which provides a consistent
set of tools for interfacing with other computers using SSH-based protocols (like scp
for copying files from one remote computer to another). Unfortunately, many common
cloud storage services do not offer an SSH based interface. Rather, they typically process
requests from clients using an HTTPS protocol. This protocol, which effectively runs the
world-wide web, is a natural choice for cloud services that most people will access
using a web browser; however, Unix does not traditionally come with a utility or command
to easily process the types of HTTPS transactions needed to network with
cloud storage. Furthermore, there must be some security when it comes to accessing
your cloud-based storage—you don’t want everyone to be able to access your files, so
your cloud service needs to have some way of authenticating people
(you and your labmates for example) that are authorized to access your data.
These problems have been overcome by a utility called rclone, the product of a
comprehensive open-source software project that brings the functionality of the
rsync utility (a common Unix tool used to synchronize and mirror file systems)
to cloud-based storage. (Note: rclone has nothing to do with the R programming
language, despite its name that looks like an R package.)
Currently rclone provides a consistent interface for accessing
files from over 35 different cloud storage providers, including Box, Dropbox, Google Drive,
and Microsoft OneDrive. Binaries for rclone can be downloaded for your desktop
machine from https://rclone.org/downloads/. We will
talk about how to install it on your HPC system later.
Once rclone is installed and in your PATH, you invoke it in your terminal
with the command rclone. Before we get into the details of the various rclone subcommands,
it will be helpful to take a glance at the information rclone records when it
configures itself to talk to your cloud service. To do so, it creates a file called ~/.config/rclone/rclone.conf, where it stores information about all the different
connections to cloud services you have set up. For example, that
file on my system looks like this:
[gdrive-rclone]
type = drive
scope = drive
root_folder_id = 1I2EDV465N5732Tx1FFAiLWOqZRJcAzUd
token = {"access_token":"bs43.94cUFOe6SjjkofZ","token_type":"Bearer","refresh_token":"1/MrtfsRoXhgc","expiry":"2019-04-29T22:51:58.148286-06:00"}
client_id = 2934793-oldk97lhld88dlkh301hd.apps.googleusercontent.com
client_secret = MMq3jdsjdjgKTGH4rNV_y-NbbGIn this configuration:
gdrive-rcloneis the name by which rclone refers to this cloud storage locationroot_folder_idis the ID of the Google Drive folder that can be thought of as the root directory ofgdrive-rclone. This ID is not the simple name of that directory on your Google Drive, rather it is the unique name given by Google Drive to that directory. You can see it by navigating in your browser to the directory you want and finding it after the last slash in the URL. For example, in the above case, the URL is:https://drive.google.com/drive/u/1/folders/1I2EDV465N5732Tx1FFAiLWOqZRJcAzUdclient_idandclient_secretare like a username and a shared secret thatrcloneuses to authenticate the user to Google Drive as who they say they are.tokenare the credentials used byrcloneto make requests of Google Drive on the basis of the user.
Note: the above does not include my real credentials, as then anyone could use them to access my Google Drive!
To set up your own configuration file to use Google Drive, you will use the rclone config
command, but before you do that, you will want to wrangle a client_id from Google. Follow
the directions at https://rclone.org/drive/#making-your-own-client-id. Things are a little different from in their step
by step, but you can muddle through to get to a screen with a client_ID and a client
secret that you can copy onto your clipboard.
Once you have done that, then run rclone config and follow the prompts. A
typical session of rclone config for Google Drive access is given
here. Don’t choose to do the advanced setup; however
do use “auto config,” which will bounce up a web page and let you authenticate rclone
to your Google account.
It is worthwhile first setting up a config file on your laptop, and making sure that it is working. After that, you can copy that config file to other remote servers you work on and immediately have the same functionality.
7.2.4.1 Encrypting your config file
While it is a powerful thing to be able to copy a config file from
one computer to the next and immediately be able to access your Google
Drive account. That might (and should) also make you a little bit
uneasy. It means that if the config file falls into the wrong hands,
whoever has it can gain access to everything on your Google Drive. Clearly
this is not good. Consequently, once you have created your rclone config
file, and well before you transfer it to another computer, you must
encrypt it. This makes sense, and fortunately it is fairly easy: you can
use rclone config and see that encryption is one of
the options. When it is encrypted, use rclone config show to see what
it looks like in clear text.
The downside of using encryption is that you have to enter your password every time you make an rclone command, but it is worth it to have the security.
Here is what it looks like when choosing to encrypt one’s config file:
% rclone config
Current remotes:
Name Type
==== ====
gdrive-rclone drive
e) Edit existing remote
n) New remote
d) Delete remote
r) Rename remote
c) Copy remote
s) Set configuration password
q) Quit config
e/n/d/r/c/s/q> s
Your configuration is not encrypted.
If you add a password, you will protect your login information to cloud services.
a) Add Password
q) Quit to main menu
a/q> a
Enter NEW configuration password:
password:
Confirm NEW configuration password:
password:
Password set
Your configuration is encrypted.
c) Change Password
u) Unencrypt configuration
q) Quit to main menu
c/u/q> q
Current remotes:
Name Type
==== ====
gdrive-rclone drive
e) Edit existing remote
n) New remote
d) Delete remote
r) Rename remote
c) Copy remote
s) Set configuration password
q) Quit config
e/n/d/r/c/s/q> qOnce that file is encrypted, you can copy it to other machines for use.
7.2.4.2 Basic Maneuvers
The syntax for use is:
The “subcommand” part tells rclone what you want to do, like copy or sync, and
the “parameter” part of the above syntax is typically a path
specification to a directory or a file. In using rclone to access the
cloud there is not a root directory, like / in Unix. Instead, each remote
cloud access point is treated as the root directory, and you refer to it
by the name of the configuration followed by a colon. In our example,
gdrive-rclone: is the root, and we don’t need to add a / after it to
start a path with it. Thus gdrive-rclone:this_dir/that_dir is a
valid path for rclone to a location on my Google Drive.
Very often when moving, copying, or syncing files, the parameters consist of:
One very important point is that, unlike the Unix commands cp and mv, rclone
likes to operate on directories, not on multiple named files.
A few key subcommands:
ls,lsd, andlslare likels,ls -dandls -l
copy: copy the contents of a source directory to a destination directory. One super cool thing about this is thatrclonewon’t re-copy files that are already on the destination and which are identical to those in the source directory.
Note that the destination directory will be created if it does not already exist.
- sync: make the contents of the destination directory look just like the
contents of the source directory. WARNING This will delete files in the destination
directory that do not appear in the source directory.
A few key options:
--dry-run: don’t actually copy, sync, or move anything. Just tell me what you would have done.--progress: give me progress information when files are being copied. This will tell you which file is being transferred, the rate at which files are being transferred, and and estimated amount of time for all the files to be transferred.--tpslimit 10: don’t make any more than 10 transactions a second with Google Drive (should always be used when transferring files)---fast-list: combine multiple transactions together. Should always be used with Google Drive, especially when handling lots of files.--drive-shared-with-me: make the “root” directory a directory that shows all of the Google Drive folders that people have shared with you. This is key for accessing folders that have been shared with you.
For example, try something like:
Important Configuration Notes!! Rather than always giving the --progress
option on the command line, or always having to remember to use
--fast-list and --tpslimit 10 (and remember what they should be…),
you can set those options to be invoked “by default” whenever you use
rclone. The developers of rclone have made this possible
by setting environment variables in your ~/.bashrc.
If you have an rclone option called --fast-limit, then the corresponding
environment variable is named RCLONE_FAST_LIMIT—basically, you
start with RCLONE_ then you just
drop the first two dashes of the option name, replace the remaining dashes
with underscores, and turn it all into uppercase to make the
environment variable. So, you should, at a minimum add these
lines to your ~/.bashrc:
7.2.4.3 filtering: Be particular about the files you transfer
rclone works a little differently than the Unix utility cp. In particular,
rclone is not set up very well to copy individual files. While there is a
an rclone command known as copyto that will allow you copy a single file,
you cannot (apparently) specify multiple, individual files that you wish to copy.
In other words, you can’t do:
In general, you will be better off using rclone to copy the contents of a directory
to the inside of the destination directory. However, there are options in rclone that
can keep you from being totally indiscriminate about the files you transfer. In other words,
you can filter the files that get transferred. You can read about that at
https://rclone.org/filtering/.
For a quick example, imagine that you have a directory called Data on you Google Drive
that contains both VCF and BAM files. You want to get only the VCF files (ending with .vcf.gz, say)
onto the current working directory on your cluster. Then something like this works:
Note that, if you are issuing this command on a Unix system in a directory
where the pattern *.vcf.gz will expand (by globbing) to multiple files, you will
get an error. In that case, wrap the pattern in a pair of single quotes to keep
the shell from expanding it, like this:
7.2.4.4 Feel free to make lots of configurations
You might want to configure a remote for each directory-specific project.
You can do that by just editing the configuration file. For example,
if I had a directory deep within my Google Drive, inside a chain of folders that
looked like, say, Projects/NGS/Species/Salmon/Chinook/CentralValley/WinterRun
where I was keeping
all my data on a project concerning winter-run Chinook salmon, then it would be
quite inconvenient to type Projects/NGS/Species/Salmon/Chinook/CentralValley/WinterRun
every time I wanted to copy or sync something within that directory. Instead,
I could add the following
lines to my configuration file, essentially copying the existing configuration and
then modifying the configuration name and the root_folder_id to be the
Google Drive identifier for the folder Projects/NGS/Species/Salmon/Chinook/CentralValley/WinterRun (which
one can find by navigating to that folder in a web browser and pulling the ID from the
end of the URL.) The updated configuration could look like:
[gdrive-winter-run]
type = drive
scope = drive
root_folder_id = 1MjOrclmP1udhxOTvLWDHFBVET1dF6CIn
token = {"access_token":"bs43.94cUFOe6SjjkofZ","token_type":"Bearer","refresh_token":"1/MrtfsRoXhgc","expiry":"2019-04-29T22:51:58.148286-06:00"}
client_id = 2934793-oldk97lhld88dlkh301hd.apps.googleusercontent.com
client_secret = MMq3jdsjdjgKTGH4rNV_y-NbbGAs long as the directory is still within the same Google Drive account, you can re-use
all the authorization information, and just change the [name] part and the root_folder_id.
Now this:
puts items into Projects/NGS/Species/Salmon/Chinook/CentralValley/WinterRun on the Google Drive
without having to type that God-awful long path name.
7.2.4.5 Installing rclone on a remote machine without sudo access
The instructions on the website require root access. You don’t have to have root
access to install rclone locally in your home directory somewhere.
Copy the download link from https://rclone.org/downloads/ for
the type of operating system your remote machine uses (most likely Linux if it is a cluster).
Then transfer that with wget, unzip it and put the binary in your PATH. It will look
something like this:
wget https://downloads.rclone.org/rclone-current-linux-amd64.zip
unzip rclone-current-linux-amd64.zip
cp rclone-current-linux-amd64/rclone ~/binYou won’t get manual pages on your system, but you can always find the docs on the web.
7.2.5 Getting files from a sequencing center
Very often sequencing centers will post all the data from a single run of a machine at a secured (or unsecured) http address. You will need to download those files to operate on them on your cluster or local machine. However some of the files available on the server will likely belong to other researchers and you don’t want to waste time downloading them.
Let’s take an example. Suppose you are sent an email from the sequencing center that says something like:
Your samples are AW_F1 (female) and AW_M1 (male). You should be able to access the data from this link provided by YCGA: http://sysg1.cs.yale.edu:3010/5lnO9bs3zfa8LOhESfsYfq3Dc/061719/
You can easily access this web address using rclone. You could set up a new
remote in your rclone config to point to http://sysg1.cs.yale.edu,
but, since you will only be using this once, to get your data, it makes
more sense to just specify the remote on the command line. This can be
done by passing rclone the URL address via the --http-url option, and
then, after that, telling it what protocol to use by adding :http: to
the command. Here is what you would use to list the directories available
at the sequencing center URL:
# here is the command
% rclone lsd --http-url http://sysg1.cs.yale.edu:3010/5lnO9bs3zfa8LOhESfsYfq3Dc/061719/ :http:
# and here is the output
-1 1969-12-31 16:00:00 -1 sjg73_fqs
-1 1969-12-31 16:00:00 -1 sjg73_supernova_fqsAha! There are two directories that might hold our sequencing data.
I wonder what is in those diretories? The rclone tree command is the
perfect way to drill down into those diretories and look at their contents:
% rclone tree --http-url http://sysg1.cs.yale.edu:3010/5lnO9bs3zfa8LOhESfsYfq3Dc/061719/ :http:
/
├── sjg73_fqs
│ ├── AW_F1
│ │ ├── AW_F1_S2_L001_I1_001.fastq.gz
│ │ ├── AW_F1_S2_L001_R1_001.fastq.gz
│ │ └── AW_F1_S2_L001_R2_001.fastq.gz
│ ├── AW_M1
│ │ ├── AW_M1_S3_L001_I1_001.fastq.gz
│ │ ├── AW_M1_S3_L001_R1_001.fastq.gz
│ │ └── AW_M1_S3_L001_R2_001.fastq.gz
│ └── ESP_A1
│ ├── ESP_A1_S1_L001_I1_001.fastq.gz
│ ├── ESP_A1_S1_L001_R1_001.fastq.gz
│ └── ESP_A1_S1_L001_R2_001.fastq.gz
└── sjg73_supernova_fqs
├── AW_F1
│ ├── AW_F1_S2_L001_I1_001.fastq.gz
│ ├── AW_F1_S2_L001_R1_001.fastq.gz
│ └── AW_F1_S2_L001_R2_001.fastq.gz
├── AW_M1
│ ├── AW_M1_S3_L001_I1_001.fastq.gz
│ ├── AW_M1_S3_L001_R1_001.fastq.gz
│ └── AW_M1_S3_L001_R2_001.fastq.gz
└── ESP_A1
├── ESP_A1_S1_L001_I1_001.fastq.gz
├── ESP_A1_S1_L001_R1_001.fastq.gz
└── ESP_A1_S1_L001_R2_001.fastq.gz
8 directories, 18 filesWhoa! That is pretty cool!. From this output we see that there are
subdirectories named AW_F1 and AW_M1 that hold the files that
we want. And, of course, the ESP_A1 samples must belong to someone
else. It would be great if we could just download the files we wanted,
excluding the ones in the ESP_A1 directories. It turns out that there is!
rclone has an --exclude option to exclude paths that match certain
patterns (see Section 7.2.4.3, above). We can
experiment by giving rclone copy the --dry-run command to see which
files will be transferred. If we don’t do any filtering, we see this
when we try to dry-run copy the directories to our local directory Alewife/fastqs:
% rclone copy --dry-run --http-url http://sysg1.cs.yale.edu:3010/5lnO9bs3zfa8LOhESfsYfq3Dc/061719/ :http: Alewife/fastqs/
2019/07/11 10:33:43 NOTICE: sjg73_fqs/ESP_A1/ESP_A1_S1_L001_I1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_fqs/ESP_A1/ESP_A1_S1_L001_R1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_fqs/ESP_A1/ESP_A1_S1_L001_R2_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_supernova_fqs/AW_M1/AW_M1_S3_L001_I1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_supernova_fqs/AW_M1/AW_M1_S3_L001_R1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_supernova_fqs/AW_M1/AW_M1_S3_L001_R2_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_supernova_fqs/AW_F1/AW_F1_S2_L001_I1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_supernova_fqs/AW_F1/AW_F1_S2_L001_R1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_supernova_fqs/AW_F1/AW_F1_S2_L001_R2_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_supernova_fqs/ESP_A1/ESP_A1_S1_L001_I1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_supernova_fqs/ESP_A1/ESP_A1_S1_L001_R1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_supernova_fqs/ESP_A1/ESP_A1_S1_L001_R2_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_fqs/AW_F1/AW_F1_S2_L001_I1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_fqs/AW_F1/AW_F1_S2_L001_R1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_fqs/AW_F1/AW_F1_S2_L001_R2_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_fqs/AW_M1/AW_M1_S3_L001_I1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_fqs/AW_M1/AW_M1_S3_L001_R1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:33:43 NOTICE: sjg73_fqs/AW_M1/AW_M1_S3_L001_R2_001.fastq.gz: Not copying as --dry-runSince we do not want to copy the ESP_A1 files we see if we can exclude
them:
% rclone copy --exclude */ESP_A1/* --dry-run --http-url http://sysg1.cs.yale.edu:3010/5lnO9bs3zfa8LOhESfsYfq3Dc/061719/ :http: Alewife/fastqs/
2019/07/11 10:37:22 NOTICE: sjg73_fqs/AW_F1/AW_F1_S2_L001_I1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:37:22 NOTICE: sjg73_fqs/AW_F1/AW_F1_S2_L001_R2_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:37:22 NOTICE: sjg73_fqs/AW_F1/AW_F1_S2_L001_R1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:37:22 NOTICE: sjg73_fqs/AW_M1/AW_M1_S3_L001_I1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:37:22 NOTICE: sjg73_fqs/AW_M1/AW_M1_S3_L001_R2_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:37:22 NOTICE: sjg73_fqs/AW_M1/AW_M1_S3_L001_R1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:37:22 NOTICE: sjg73_supernova_fqs/AW_F1/AW_F1_S2_L001_I1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:37:22 NOTICE: sjg73_supernova_fqs/AW_F1/AW_F1_S2_L001_R2_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:37:22 NOTICE: sjg73_supernova_fqs/AW_F1/AW_F1_S2_L001_R1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:37:22 NOTICE: sjg73_supernova_fqs/AW_M1/AW_M1_S3_L001_I1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:37:22 NOTICE: sjg73_supernova_fqs/AW_M1/AW_M1_S3_L001_R1_001.fastq.gz: Not copying as --dry-run
2019/07/11 10:37:22 NOTICE: sjg73_supernova_fqs/AW_M1/AW_M1_S3_L001_R2_001.fastq.gz: Not copying as --dry-runBooyah! That gets us just what we want. So, then we remove the --dry-run option,
and maybe add -v -P to give us verbose output and progress information, and copy all of our files:
7.3 Editing Files on a Remote Server using a Local Text Editor
Section 7.7 discusses the value of getting good at using a text-based
text editor like vim or emacs, or even the easy-to-use nano. That is
all well and good; however, if you have become proficient with a local
(i.e., running on your laptop) text editor with powerful features and
outstanding syntax highlighting, like SublimeText, which
is available for Mac, Windows, and Linux, then it can be very nice to
be able to directly edit files on your remote server or cluster using your
laptop’s own installation of SublimeText.
It turns out that this is possible through the miracle of SSH port forwarding. Briefly, it works like this:
- When you log into your server with
sshyou tell the server to connect a remote port on the server to a local port on your laptop. This way, the server can send additional data streams back and forth through those connected ports to you. - Then on the server, you run a shell script called
rmatethat will can open a file on the server and send its contents out through the remote port. Your laptop picks up these contents on the local port and can send those contents to SublimeText using a SublimeText plugin called RemoteSubl. - Editing the contents of that file
with SublimeText feels just like editing a local file on your laptop, but when you
save your edits, SublimeText sends the changes back out through the local port to the
remote port on your server, where
rmateapplies the changes to the file on the server.
It is a great system for editing things on the remote server if you are familiar with SublimeText (which you should get familiar with, because it is a great editor!).
More detailed, step-by-step instructions on how to set this up follow.
7.3.1 Step 1: Set up your SSH config file to automatically apply port forwarding to your connections to your server
The first thing we will do is something that can especially useful if you have several servers you connect to, and you would like to have shorter names for accessing them: set up an “alias” to them in your SSH config file. Here we show how to set up such an alias to the SUMMIT cluster at Boulder.
To do so, edit your ~/.ssh/config file by adding the following lines to it:
Host summit
HostName login11.rc.colorado.edu
RemoteForward 52XXX localhost:52698
But change the XXX in 52XXX to three digits of your choice. This is important
so you aren’t trying to access the same port as another user at the same time. For
example, use the digits of your birthday, like 52122 if you were born on January 22, etc.
This step connects port number 52XXX on the server to the local port 52698 on your laptop.
Note that the login node you choose could be login12.rc.colorado.edu rather
than login11.rc.colorado.edu, or even some other number if you routinely login
to a specific login node (for example to use tmux…see Section 7.4).
Once you have done that, you should logout of SUMMIT, and then, when you log back in in the future you can use
instead of, for example,
and, when it logs you in, it will enable the port forwarding.
7.3.2 Download SublimeText and add the RemoteSubl package to it
If you haven’t already downloaded SublimeText, you should do that and you should experiment with using it. It is an outstanding text editor. You can try it for free for a long time (indefinitely, it seems), but if you find you like it, then officially buying a license for it is is a good idea.
Once you have it installed, you must install the RemoteSubl plugin. This is done
with SublimeText’s package control system. The steps to do this are:
Hit Shift-Command-P on a mac. On windows I think it is Shift-Windows-P. When you do this, sublime text will open a little text window on your screen. Type into that window:
Package Control: Install Package. You don’t have to type very much of that phrase before you see the whole phrase as a possible completion below where you are typing. When you see the full phrase, use the arrow keys to select that phrase and hit Return. If you don’t seePackage Control: Install Packageyou might seeInstall Package Control. Select that and install Package Control. After that,Package Control: Install Packageshould show up.This should give you another text box. Start typing
RemoteSublinto that window, until you see it in the possible completions. Select it from the completions (with your arrow keys, for example) and then hit return.
7.3.3 Download the rmate shell script to your server and put it on your PATH
On the server, we need the rmate command to send the contents of a file
you wish to edit to the remote port that will get forwarded to the local port
on your laptop. This command is a shell script that you can download with
wget.
As we talked about in an earlier section, you should have a directory,
~/bin that is in your PATH, because you should have a line in your
~/.bashrc file like:
If this is the case, then simply cd-ing to ~/bin on SUMMIT and running the commands:
should get you rmate and make it executable.
7.3.4 Using rmate
If everything has gone according to plan, then, if you have logged into summit
using the summit alias (i.e. using ssh username@summit), then you should
be able to edit any file on your server with:
where path/to/file represents the path to whatever file you want to
edit, and 52XXX is actually the number of the remote port you are forwarding
from, as set up in your ~/.ssh/config file. For example, in keeping with
the example above, this would be:
to edit the .bashrc file on your remote server, or,
etc.
Note that, by default, rmate will open a new tab in the currently active
SublimeText window. If you want to open the file in its own window, you
can use:
The -n option forces the opening of a new SublimeText window.
If you want to edit multiple files at once, for example, all the
files in a scripts folder on your remote machine, you can do like this:
If you have a lot of files in that folder, then keeping track of them
in SublimeText can be made a lot easier if you choose
View->Side Bar->Show Open Files from the SublimeText menu options.
This will show the names of all the open files in a side bar to the left.
Because it can be a hassle to remember your remote port number and type
it each time you use rmate, you can set up an alias in the
~/.bashrc file on your server by adding the line:
Then, on the command line, you can simply use
and open remote files on your local SublimeText.
When you have edited the file, save the change in SublimeText and then close the window. It is that easy.
Note that if you lose connection to the server (for example you close your laptop and it goes to sleep), then you will get a message telling you that SublimeText is no longer connected to any files on the server, and you will have to reconnect them if you want to edit them.
7.4 tmux: the terminal multiplexer
Many universities have recently implemented a two-factor
authentication requirement for access to their computing resources
(like remote servers and clusters). This means that every time
you login to a server on campus (using ssh for example) you must
type your password, and also fiddle with your phone. Such systems
preclude the use of public/private key pairs that historically allowed
you to access a server from a trusted client (i.e., your own secured
laptop) without having to type in a password. As a consequence, today,
opening multiple sessions on a server using ssh and two-factor
authentication requires a ridiculous amount of additional typing and
phone-fiddling, and is a huge hassle. But, when working on a remote
server it is often very convenient to have multiple separate shells that
you are working on and can quickly switch between.
At the same time. When you are working on the shell of a remote machine
and your network connection goes down, then, typically the bash session on
your remote machine will be forcibly quit, killing any jobs that you might
have been in the middle of (however, this is not the case if you submitted
those jobs through a job scheduler like SLURM. Much more on that in the
next chapter.). And, finally, in a traditional
ssh session to a remote machine, when you close your laptop, or put it
to sleep, or quit the Terminal application, all of your active bash sessions
on the remote machine will get shut down. Consequently, the next time
you want to work on that project, after you have logged
onto that remote machine you will have to go through the laborious steps
of navigating to your desired working directory, starting up any processes
that might have gotten killed, and generally getting yourself
set up to work again. That is a serious buzz kill!
Fortunately, there is an awesome utility called tmux, which is short for
“terminal multiplexer” that solves most of the problems we just described.
tmux is similar in function to a utility called screen, but it is easier
to use while at the same time being more customizable and configurable
(in my opinion). tmux is basically your ticket to working way more
efficiently on remote computers, while at the same time looking
(to friends and colleagues, at least) like
the full-on, bad-ass Unix user.
In full confession, I didn’t actually start using tmux until some
five years after a speaker at a workshop delivered an incredibly
enthusiastic presentation about tmux and how much he was in love
with it. In somewhat the same fashion that I didn’t adopt RStudio shortly
after its release, because I had my own R workflows that I had hacked
together myself, I thought to myself: “I have public/private key pairs
so it is super easy for me to just start another terminal window and login
to the server for a new session. Why would I need tmux?” I also didn’t
quite understand how tmux worked initially: I thought that I had to
run tmux simultaneously on my laptop and on the server, and that those
two processes would talk to one another. That is not the case! You
just have to run tmux on the server and all will work fine!
The upshot of that confession is that you should not be a bozo like me,
and you should learn to use tmux right now! You will thank yourself
for it many times over down the road.
7.4.1 An analogy for how tmux works
Imagine that the first time you log in to your remote server you also have the option of speaking on the phone to a super efficient IT guy who has a desk in the server room. This dude never takes a break, but sits at his desk 24/7. He probably has mustard stains on his dingy white T-shirt from eating ham sandwiches non-stop while he works super hard. This guy is Tmux.
When you first speak to this guy after logging in, you have to preface your
commands with tmux (as in, “Hey Tmux!”). He is there to help you
manage different terminal windows with different bash shells or
processes going on in them. In fact, you can think of it this way: you can
ask him to set up
a terminal (i.e., like a monitor), right there on his desk, and then create
a bunch of windows on that terminal for you—each
one with its own bash shell—without having to do a separate login for
each one. He has created all those windows, but you still get to use them.
It is like he has a miracle-mirroring device that lets you operate
the windows that are on the terminal he set up for you on his desk.
When you are done working on all those windows, you can tell Tmux that you want to detach from the special terminal he set up for you at the server. In response he says, “Cool!” and shuts down his miracle-mirroring device, so you no longer see those different windows. However, he does not shut down the terminal on his desk that he set up for you. That terminal stays on, and any of your processes happening on it keep chugging away…even after you logout from the server entirely, throw the lid down on your laptop, have drinks with your friends at Social, downtown, watch an episode of Parks and Rec, and then get a good night’s sleep.
All through the night, Tmux is munching ham sandwiches and keeping an eye on that terminal he set up for you. When you log back onto the server in the morning, you can say “Hey Tmux! I want to attach back to that terminal you set up for me.” He says, “No problem!”, turns his miracle-mirroring device back on, and in an instant you have all of the windows on that terminal back on your laptop with all the processes still running in them—in all the same working directories—just as you left it all (except that if you were running jobs in those windows, some of those jobs might already be done!).
Not only that, but, further, if you are working on the server when a local thunderstorm fries the motherboard on your laptop, you can get a new laptop, log back into the server and ask Tmux to reconnect you to that terminal and get back to all of those windows and jobs, etc. as if you didn’t get zapped. The same goes for the case of a backhoe operator accidentally digging up the fiber optic cable in your yard. Your network connection can go down completely. But, when you get it up and running again, you can say “Hey Tmux! Hook me up!” and he’ll say, “No problem!” and reconnect you to all those windows you had open on the server.
Finally, when you are done with all the windows and jobs on the terminal that Tmux set up for you, you can ask him to kill it, and he will shut it down, unplug it, and, straight out of Office Space, chuck it out the window. But he will gladly install a new one if you want to start another session with him.
That dude is super helpful!
7.4.2 First steps with tmux
The first thing you want to do to make sure Tmux is ready to help you is to simply type:
This should return something like:
/usr/bin/tmuxIf, instead, you get a response like tmux: Command not found. then tmux
is apparently not installed
on your remote server, so you
will have to install it yourself, or beg your sysadmin to do so (we will cover
that in a later chapter). If you
are working on the Summit supercomptuer in Colorado or on Hummingbird at
UCSC, then tmux is installed already. (As of Feb 16, 2020, tmux was
not installed on the Sedna cluster at the NWFSC, but I will request that it
be installed.)
In the analogy, above, we talked about Tmux setting up a terminal
in the server room. In tmux parlance, such a “terminal” is called
a session. In order to be able to tell Tmux that you want to reconnect
to a session, you
will always want to name your sessions so you will request a new
session with this syntax:
You can think of the -s as being short for “session.” So it is basically a short
way of saying, “Hey Tmux, give me a new session named froggies.”
That creates a new session called froggies, and you can imagine we’ve
named it that because we will use it for work on a frog genomics project.
The effect of this is like Tmux firing up a new terminal in his server room, making a window on it for you, starting a new bash shell in that window, and then giving you control of this new terminal. In other words, it is sort of like he has opened a new shell window on a terminal for you, and is letting you see and use it on your computer at the same time.
One very cool thing about this is that you just got a new bash shell without having to login with your password and two-factor authentication again. That much is cool in itself, but is only the beginning.
The new window that you get looks a little different. For one thing, it has a section, one line tall, that is green (by default) on the bottom. In our case, on the left side it gives the name of the session (in square brackets) and then the name of the current window within that session. On the right side you see the hostname (the name of the remote computer you are working on) in quotes, followed by the date and time. The contents in that green band will look something like:
[froggies] 0:bash* "login11" 20:02 15-Feb-20tmux can spawn for you.login11). Many clusters have multiple login
or head nodes, as they are called. The next time you login to the cluster, you
might be assigned to a different login node which will have no idea about your
tmux sessions. If that were the case in this example I would have to use slogin login11 and
authenticate again to get logged into login11 to reconnect to my
tmux session, froggies. Or, if you were a CSU student and wanted to login specifically to
the login11 node on Summit the next time you logged on you could do
ssh username@colostate.edu@login11.rc.colorado.edu. Note the specific login11 in that
statement.
Now, imagine that we want to use this window in our froggies session, to look at some
frog data we have. Accordingly, we might navigate to the directory where those data live
and look at the data with head and less, etc. That is all great, until we realize that
we also want to edit some scripts that we wrote for processing our froggy data. These scripts
might be in a directory far removed from the data directory we are currently in, and we don’t really
want to keep navigating back and forth between those two directories within a single bash shell.
Clearly, we would like to have two windows that we could switch between: one for inspecting
our data, and the other for editing our scripts.
We are in luck! We can do this with tmux. However, now that we are safely working in a session
that tmux started for us, we no longer have to shout “Hey Tmux!” Rather we can just “ring a little
bell” to get his attention. In the default tmux configuration, you do that by pressing
<cntrl>-b from anywhere within a tmux window. This is easy to remember because it is like
a “b” for the “bell” that we ring to get our faithful servant’s attention. <cntrl>-b is known
as the “prefix” sequence that starts all requests to tmux from within a session.
The first thing that we are going to do is ask tmux to let us assign a more descriptive,
name—data to be specific—to the current window. We do this with
<cntrl>-b ,(That’s right! It’s a control-b and then a comma. tmux likes to get by on a minimum number
of keystrokes.) When you do that, the green band at the bottom of the window changes color
and tells you that you can rename the current window. We simply use our keyboard to
change the name to “data”. That was super easy!
Now, to make a new window with a new bash shell that we can use for writing scripts
we do <cntrl>-b c. Try it! That gives you a new window within the froggies session
and switches your focus to it. It is as if Tmux (in his mustard-stained shirt) has created
a new window on the froggies terminal, brought it to the front, and shared it with you.
The left side of the green tmux status bar at the bottom of the screen now says:
[froggies] 0:data- 1:bash*Holy Moly! This is telling you that the froggies session has two windows in it: the first
numbered 0 and named data, and the second numbered 1 and named bash. The - at the end
of 0:data- is telling you that data is the window you were previously focused on, but that
now you are currently focused on the window with the * after its name: 1:bash*.
So, the name bash is not as informative as it could be. Since we will be using this
new window for editing scripts, let’s rename it to edit. You can do that with
<cntrl>-b ,. Do it!
OK! Now, if you have been paying attention, you probably realize that tmux has given us
two windows (with two different bash shells) in this session called froggies. Not only that
but it has associated a single-digit number with each window. If you are all about keyboard
shortcuts, then you probably have already imagined that tmux will let you switch between
these two windows with <cntrl>-b plus a digit (0 or 1 in this case). Play with that.
Do <cntrl>-b 0 and <cntrl>-b 1 and feel the power!
Now, for fun, imagine that we want to have another window and a bash shell for launching
jobs. Make a new window, name it launch, and then switch between those three windows.
Finally. When you are done with all that, you tell Tmux to detach from this session by typing:
<cntrl>-b d(The d is for “detach”). This should kick you back to the shell from which you
first shouted “Hey Tmux!” by issuing the tmux a -t froggies command. So, you
can’t see the windows of your froggies session any longer, but do not despair!
Those windows are still on the monitor Tmux set up for you, casting an eerie glow
on his mustard stained shirt.
If you want to get back in the driver’s seat with all of those windows, you simply need to
tell Tmux that you want to be attached again via his miracle-mirroring device. Since we
are no longer in a tmux window, we don’t use our <cntrl-b> bell to get Tmux’s attention.
We have to shout:
The -t flag stands for “target.” The froggies session is the target of our
attach request. Note that if you don’t like typing that much, you can shorten this to:
Of course, sometimes, when you log back onto the server, you won’t remember the name
of the tmux session(s) you started. Use this command to list them all:
The ls here stands for “list-sessions.” This can be particularly useful if you
actually have multiple sessions. For example, suppose you are a poly-taxa genomicist,
with projects not only on a frog species, but also on a fish and a bird species. You
might have a separate session for each of those, so that when you issue tmux ls the
result could look something like:
% tmux ls
birdies: 4 windows (created Sun Feb 16 07:23:30 2020) [203x59]
fishies: 2 windows (created Sun Feb 16 07:23:55 2020) [203x59]
froggies: 3 windows (created Sun Feb 16 07:22:36 2020) [203x59]That is enough to remind you of which session you might wish to reattach to.
Finally, if you are all done with a tmux session, and you have detached from it,
then from your shell prompt (not within a tmux session) you can do, for example:
to kill the session. There are other ways to kill sessions while you are in them, but that is not so much needed.
Table 7.1 reviews the minimal set of
tmux commands just described. Though there is much more that
can be done with tmux, those commands will get you started.
| Within tmux? | Command | Effect |
|---|---|---|
| N | tmux ls |
List any tmux sessions the server knows about |
| N | tmux new -s name |
Create a new tmux session named “name” |
| N | tmux attach -t name |
Attach to the existing tmux session “name” |
| N | tmux a -t name |
Same as “attach” but shorter. |
| N | tmux kill-session -t name |
Kill the tmux session named “name” |
| Y | <cntrl>-b , |
Edit the name of the current window |
| Y | <cntrl>-b c |
Create a new window |
| Y | <cntrl>-b 3 |
Move focus to window 3 |
| Y | <cntrl>-b & |
Kill current window |
| Y | <cntrl>-b d |
Detach from current session |
| Y | <cntrl>-l |
Clear screen current window |
7.4.3 Further steps with tmux
The previous section merely scratched the surface of what is possible with tmux.
Indeed, that is the case with this section. But here I just want to leave you with a
taste for how to configure tmux to your liking, and also with the ability to create
different panes within a window within a session. You guessed it! A pane is made by
splitting a window (which is itself a part of a session) into two different
sections, each one running its own bash shell.
Before we start making panes, we set some configurations that make the
establishment of panes more intuitive (by using keystrokes that are easier
to remember) and others that make it easier to quickly adjust the size of the panes.
So, first, add these lines to ~/.tmux.conf:
# splitting panes
bind \ split-window -h -c '#{pane_current_path}'
bind - split-window -v -c '#{pane_current_path}'
# easily resize panes with <C-b> + one of j, k, h, l
bind-key j resize-pane -D 10
bind-key k resize-pane -U 10
bind-key h resize-pane -L 10
bind-key l resize-pane -R 10Once you have updated ~/.tmux.conf you need to reload that
configuration file in tmux. So, from within a tmux session,
you do <cntrl>-b :. This let’s you type a tmux command in the lower
left (where the cursor has become active). Type source-file ~/.tmux.conf
The comments show what each line is intended to do, and you
can see that the configuration “language” for tmux is relatively
unintimidating. In plain language, these configurations are saying that, after this
configuration is made active, <cntrl>-b / will split a window (or a pane),
vertically, in to two panes. (Note that this is easy to remember
because on an American keyboard, the \ and the |, share a key. The latter
looks like a vertical separator, and would thus be a good key stroke
to split a screen vertically, but why force ourselves to hit the shift key as well?).
Likewise, <cntrl>-b - will split a window (or a pane) into two panes.
tmux
window with four panes. The two vertical ones on the left show a yaml
file and a shell script being edited in vim, and the remaining two
house shells for looking at files in two different directories.
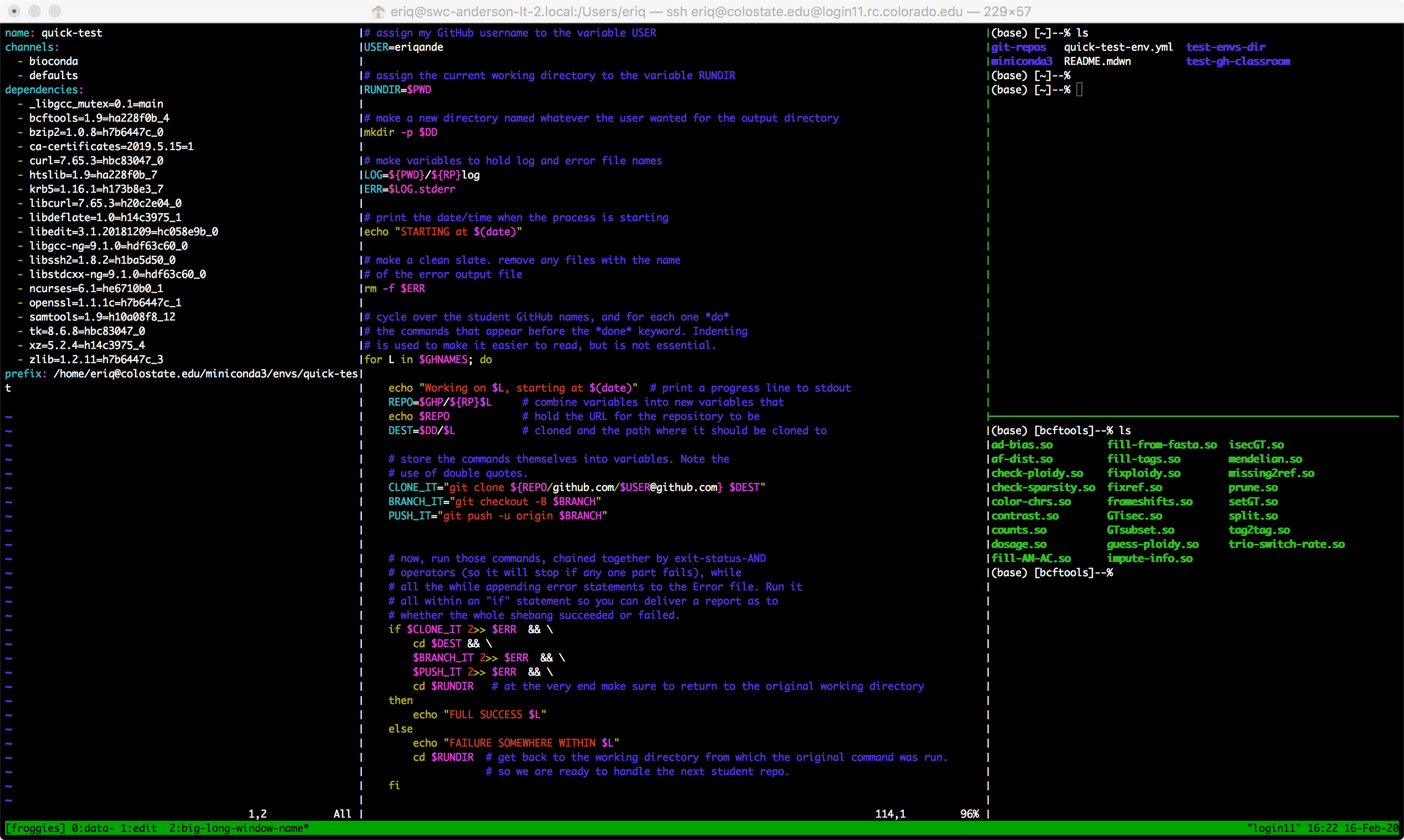
FIGURE 7.7: A tmux window with four panes.
This provides almost endless opportunities for customizing the appearance of your terminal workspace on a remote machine for maximum efficiency. Of course, doing so requires you know a few more keystrokes for handling panes. These are summarized in Table 7.2.
| Within tmux? | Command | Effect |
|---|---|---|
| Y | <cntrl>-b / |
Split current window/pane vertically into two panes |
| Y | <cntrl>-b - |
Split current window/pane horizontally into two panes |
| Y | <cntrl>-b arrow |
Use <cntrl>-b + an arrow key
to move sequentially amongst
panes |
| Y | <cntrl>-b x |
Kill current the current pane |
| Y | <cntrl>-b q |
Paint big ID numbers (from 0 up) on the panes for a few seconds. Hitting a number before it disappears moves focus to that pane. |
| Y | <cntrl>-b [hjkl] |
Resize the current pane, h = Left, j = Down, k = Up, l = Right. It takes a while to understand which boundary will move. |
| Y | <cntrl>-b z |
Zoom current pane to full
size. <cntrl>-b z again
restores it to original size. |
Now that you have seen all these keystrokes, use <cntrl>-b \ and <cntrl>-b - to split your windows
up into a few panes and try them out. It takes a while to get used to it, but once you get
the hang of it, it’s quite nice.
7.5 tmux for Mac users
I put this in an entirely different section, because, if you are comfortable in
Mac-world, already, working with tmux by way of the extraordinary Mac application
iTerm2 feels like home and it is a completely different experience than
working in tmux the way we have, so far.
iTerm2 is a sort of fully customizable and way better replacement for the standard Mac Terminal application. It can be downloaded for free from its web page https://www.iterm2.com/. You can donate to the project there as well. If you find that you really like iTerm2, I recommend a donation to support the developers.
There are far too many features in iTerm2 to cover here, but I just want to describe
one very important feature: iTerm2 integration with tmux. If you have survived the
last section, and have gotten comfortable with hitting <cntrl>-b and then a series
of different letters to effect various changes, then that is a good thing, and will
serve you well. However, as you continue your journey with tmux, you may have found that
you can’t scroll up through the screen the way you might be used to when working in Terminal
on a Mac. Further, you may have discovered that copying text from the screen, when you
finally figured out how to scroll up in it, involves a series of emacs-like keystrokes.
This is fine if you are up for it, but it is understandable that a Mac user might yearn
for a more Mac-like experience. Fortunately, the developers of iTerm2 have made your tmux experience
much better! They exploit tmux’s -CC option, which puts tmux into “control mode” such that
iTerm2 can send its own simple text commands to control tmux, rather than the user sending
commands prefaced by <cntrl>-b. The consequence of this is that iTerm2 has a series of menu options
for doing tmux actions, and all of these have keyboard shortcuts that seem more natural to
a Mac user. You can establish sessions, open new windows (as tabs in iTerm, if desired) and
even carve windows up into multiple panels—all from a Mac-style interface that is quite forgiving
in case you happen to forget the exact key sequence to do something in tmux.
Finally, using tmux via iTerm2 you get mouse interaction like you expect: you can use the
mouse to select different panes and move the dividers between them, and you can scroll back
and select text with the mouse, if desired.
On top of that, iTerm2 has great support for creating different profiles that you can assign to different remote servers. These can be customized with different login actions (including storage of remote server passwords in the Apple keychain, so you don’t have to type in your long, complex passwords every time you log in to a remote server you can’t set a public/private keypair on), and with different color schemes that can help you to keep various windows attached to various remote servers straight.
You can read about it all at the iTerm2 website. I will just add that using
iTerm with tmux might require a later version of tmux than is installed
on your remote server or cluster. I recommend that you use mamba (see Section 7.6.2)
to install the latest version of tmux into a new tmux environment.
So, there are four major steps to getting this all set up. 1) Install a sufficiently
new version of tmux on your server (i.e. on summit, or alpine, or sedna, etc.), and then
2) set up an ssh host alias in your ssh config file, 3) save your password in iTerm2’s
keychain-encrypted password storage, and 4) set up a profile in iTerm that will orchestrate connecting to your server via
tmux with -CC option.
Each of those four steps is covered in the next four sections.
7.5.1 Getting a newer version of tmux on the server
Because your server likely comes with an old version of tmux, we will use mamba to install the latest version for your own use. You do these steps while logged on to your remove server.
If you have previously used tmux, make sure that you have no tmux sessions running. Do this by detaching from any current tmux session (if any) with
<cntrl>-b d. Then, when you just have a normal shell (not throughtmux), dotmux lsto see if there are any othertmuxsessions. If there are, then you should do:tmux kill-session -t namewithnamebeing seing to each of the session names. Or, more simply, and directly, just do:tmux kill-server. After that, check withtmux lsto make sure no othertmuxsessions are running. (If you have never used tmux, then you should not have to do any of those preceding steps!)Install the latest version of tmux into its own environment, using
mamba:
Once that is done, you can activate the tmux session and then print the absolute path to the tmux binary:
The last command should give you an absolute path to your new tmux binary.
Copy that and paste it into a text file for later use. We will call that
path tmux-absolute-path.
On Alpine for me, the path will be something like: /projects/eriq@colostate.edu/miniforge3/tmux/bin/tmux.
It might be somewhat different for you. No worries, so long as
which tmux returns a path when your tmux environment is activated you should
be good to go.
When you are done with recording the absolute path to tmux, go ahead and deactivate the tmux environment:
7.5.2 Making an ssh host alias to your server
This is done on your own shell on your laptop! The SSH utility accepts
various configurations in a file that is at ~/.ssh/config. We are going to
add the address of our server to this config file to shorten the command we
type to login to the server.
For example, if we need to log into a computer whose address is:
login11.rc.colorado.eduThen we can add a section to our .ssh/config file that looks like this:
Host rclog
HostName login11.rc.colorado.edu
That gives us an alias named rclog that the ssh utility will recognize as an
alias to the server login11.rc.colorado.edu, so that you could login to
that server with ssh username@rclog.
So, go ahead and add such lines to your ~/.ssh/config file using nano.
7.5.3 Saving a password in iTerm
You do this in the iTerm2 app on your laptop.
You must populate iTerm’s password manager with the password that you usee to your remove server. This stores your password in an encrypted file and then iTerm will be able to provide it when you log in to the remove server. To do this:
- Choose
Window->Password Managerfrom iTerm’s menu. Note that “Password Manager” is down near the bottom. - Hit the “+” to add a new password. Doing so will pop up a “New Account” line. Double click “New Account” to edit that and change it to the name of your server (just so that you know what that password goes to).
- When
the line for your server is highlighted, click “Edit Password”. In the popup,
put your password for the server in there. If you are doing this for Alpine,
you need to type your password in with the trailing
,push. Then hit “Close”.
7.5.4 Adding a profile in iTerm that uses tmux
From iTerm2’s menu, choose,
Profiles->Open Profiles. Then click theEdit Profilesbutton. At the bottom left of that window hit the plus symbol to add a new profile. The picture of this is showing that you could assing a name liketmux-summitto the profile, but you should go ahead and name it whatever you might find appropriate.
Add a Command that is of the form
ssh -t username@host "tmux-absolute-path -CC attach -t iterm || tmux-absolute-path -CC new -s iterm"to the text box left of the dropdown menu where you can find the “Command”.
Note that you have to change username and host to appropriate values for
youself. The host should the the ssh alias that you set up above an the
tmux-absolute-path should be what you copied down previously. So, my whole command might
look something like:
ssh -t eriq@colostate.edu@rclog "/projects/eriq@colostate.edu/miniforge3/tmux/bin/tmux -CC attach -t iterm || /projects/eriq@colostate.edu/miniforge3/tmux/bin/tmux -CC new -s iterm"It is probably best to edit this whole command in a text editor, customizing it with
your username and path and then copy it en masse into the “Command” window in the
iTerm profile. Here is another picture:
A little background is in order here. What this command says is:
“login to the remote server via the alias that we set up in the ~/.ssh/config, and try to
attach to a tmux session named iterm. If attaching to session iterm fails (because
there is no tmux session named iterm) then create a new tmux session named iterm.”
Note that the tmux session does not have to be named iterm but it is handy when it is,
because then you know if you do tmux ls that that session is used by the iTerm2 application.
- Now, we set this iTerm profile up to open the iTerm password manager
when it sees the phrase
Password:when it logs into the remote server. (Note that if your server has a different password prompt that would not be detected by the regular expressionPassword:, then you should modify that regular expression entered below, accordingly).
Choose the “Advanced” tab on the upper right and “Edit” the triggers:
Once you have done that, click the “+” to add a trigger:
 Then, in the following fields do:
Then, in the following fields do:
- Regular Expression: Add
Password: - Action: Choose “Open Password Manager”
- Parameters: Choose the name given to the password from the dropdown menu
- Instant: Make sure it is checked
- Enabled: Make sure it is checked
You might have to extend the area of the screen a little bit.
 When you are done. Hit “Close”
When you are done. Hit “Close”
7.5.5 Using the iTerm-with-tmux profile to connect, disconnect, and reconnect to sessions
There are a couple of other settings that are nice to set. See the end of the the video Setting up tmux integration with iTerm2 to access Alpine or any other remote server for some suggestions.
7.6 Installing Software on an HPCC
In order to do anything useful on a remote computer or a high-performance computing cluster (called a “cluster” or an “HPCC”) you will need to have software programs for analyzing data. As we have seen, a lot of the nuts and bolts of writing command lines uses utilities that are found on every Unix computer. However almost always your bioinformatic analyses will require programs or software that do not come “standard” with Unix. For example, the specialized programs for sequence assembly and alignment will have to be installed on the cluster in order for you to be able to use them.
It turns out that installing software on a Unix machine (or cluster) has not always been a particularly easy thing to do for a number or reasons. First, for a long time, Unix software was largely distributed in the form of source code: the actual programming code (text) written by the developers that describes the actions that a program takes. Such computer code cannot be run directly, it first must be compiled into a binary or executable program. Doing this can be a challenging process. First, computer code compilation can be very time consuming (if you use R on Linux, and install all your packages from CRAN—which requires compilation—you will know that!). Secondly, vexing errors and failures can occur when the compiler or the computer architecture is in conflict with the program code. (I have lost entire days trying to solve compiling problems). On top of that, in order to run, most programs do not operate in a standalone fashion; rather, while a program is running, it typically depends on computer code and routines that must be stored in separate libraries on your Unix computer. These libraries are known as program dependencies. So, installing a program requires not just installing the program itself, but also ensuring that the program’s dependencies are installed and that the program knows where they are installed. As if that were not enough, the dependencies of some programs can conflict (be incompatible) with the dependencies of other programs, and particular versions of a program might require particular versions of the dependencies. Additionally, some versions of some programs might not work with particular versions (types of chips) of some computer systems. Finally, most systems for installing software that were in place on Unix machines a decade ago required that whoever was installing software have administrative privileges on the computer. On an HPCC, none of the typical users have administrative privileges which are, as you might guess, reserved for the system administrators.
For all the reasons above, installing software on an HPCC used to be a harrowing affair: you either had to be fluent in compilers and libraries to do it yourself in your home directory or you had to beg your system administrator. (Though our cluster computing sysadmins at NMFS are wonderful, that is not always the case…see Dilbert). On HPCC’s the system administrators have to contend with requests from multiple users for different software and different versions. They solve this (somewhat headachey) problem by installing software into separate “compartments” that allow different software and versions to be maintained on the system without all of it being accessible at once. Doing so, they create modules of software. This is discussed in the following section.
For over a decade however, a large group of motivated people have worked on creating and updating software management system that can be quite useful for getting software installed on a cluster. The project started in 2012 and was known as Anaconda. It was a python-based software distribution system for Python and R environments for data science. Subsequently, it was realized that this system would be good for distributing many other software packages, and a separate project called Miniconda was spun off from Anaconda. The Miniconda approach tries to solve many of the problems encountered in maintaining software on a computer system. First, Miniconda maintains a huge repository of programs that are already pre-compiled for a number of different chip architectures, so that programs can usually be installed without the time-consuming compiling process. Second, the repository maintains critical information on the dependencies for each software program, and about conflicts and incompatibilities between different versions of programs, architectures and dependencies. Third, the Miniconda system is built from the ground up to make it easy to maintain separate software environments on your system. These different environments have different software programs or different versions of different software programs. Such an approach was originally used so developers could use a single computer to test any new code they had written in a number of different computing environments; however, it has become an incredibly valuable tool for ensuring that your analyses are reproducible: you can give people not just the data and scripts that you used for the analysis, but also the computing/software environment (with all the same software versions) that you used for the analysis. And, finally, all of this can be done with Miniconda without having administrative privileges. Effectively, Miniconda manages all these software programs and dependencies within your home directory. Section 7.6.2 provides details about Miniconda and describes how to use it to install bioinformatics software.
Before we proceed, we offer one last word about Miniconda. Miniconda was originally implemented
in Python; however the computing necessary to maintain version compatibility in the dependencies
for certain software environments can be substantial. Eventually the standard conda command
from Miniconda was too slow to be useful for certain software packages (I remember waiting hours
for it to install MultiQC—a common Java based bioinformatic tool.) Accordingly, almost all of the
functionality of Miniconda was reimplemented in much faster, compiled C++ code in a project called
mamba. (Since these all grew out of Python, people love their snake names! And they are appropriate—the
package acquisition commands in conda run about as fast as you would expect an overly large anaconda
that had just eaten a cow to move. By comparison, package acquisition using mamba is lightning fast, just
as one would expect from a long, svelte, black or green mamba). So, when you see conda and mamba,
just know that they do effectively the same things, except that mamba is way faster and is
essentially required for effective use of the Miniconda package management system for bioinformatics.
7.6.1 Modules
The easiest way to install software on a remote computer or HPCC is to have someone
else do it! On HPCCs it is common for the system administrators to install software into
different “compartments” using the module utility. This allows for a large number of
different software packages to be “pre-installed” on the computer, but the software
is not accessible/usable until the user explicitly asks for the software to be made
available in a shell. Users ask for software to be made available in the shell
with the module load modulefile command. The main action of such a command is to
modify the user’s PATH variable to include the software’s location. (Sometimes, additional
shell environment variables are set). By managing software in this way, system administrators
can keep dependency conflicts
between different software programs that are seldom used together from causing problems.
If you work on an HPCC with administrators who are attuned to people doing bioinformatic
work, then all the software you might need could already be available using in modules. To
see what software is available you can use module avail. For example, on the SEDNA cluster
which was developed for genomic research, module avail shows a wide range of different
software specific to sequence assembly, alignment, and analysis:
% module avail
------------------------- /usr/share/Modules/modulefiles --------------------------
dot module-git module-info modules null use.own
-------------------------------- /act/modulefiles ---------------------------------
impi mvapich2-2.2/gcc openmpi-1.8/gcc openmpi-3.0.1/gcc
intel mvapich2-2.2/intel openmpi-1.8/intel openmpi-3.0.1/intel
mpich/gcc openmpi-1.6/gcc openmpi-2.1.3/gcc
mpich/intel openmpi-1.6/intel openmpi-2.1.3/intel
------------------------- /opt/bioinformatics/modulefiles -------------------------
aligners/bowtie2/2.3.5.1 bio/fastqc/0.11.9 bio/stacks/2.5
aligners/bwa/0.7.17 bio/gatk/4.1.5.0 compilers/gcc/4.9.4
assemblers/trinity/2.9.1 bio/hmmer/3.2.1 compilers/gcc/8.3.0
bio/angsd/0.931 bio/jellyfish/2.3.0 lib64/mpc-1.1.0
bio/augustus/3.2.3 bio/mothur/1.43.0 R/3.6.2
bio/bamtools/2.5.1 bio/picard/2.22.0 tools/cmake/3.16.4
bio/bcftools/1.10.2 bio/prodigal/2.6.3 tools/pigz/2.4
bio/blast/2.10.0+ bio/salmon/1.1.0
bio/blast/2.2.31+ bio/samtools/1.10Most of the bioinformatics tools are stored in the directory
/opt/bioinformatics/modulefiles, which is not a standard storage location for
modules, so, if you are using SEDNA, and you want to use these modules,
you must include that path in the MODULEPATH shell environment variable.
This can be done by updating the MODULEPATH in your ~/.bashrc file, adding
the line:
Once that is accomplished, every time you open a new shell, your MODULEPATH will be
set appropriately.
If you work on an HPCC that is not heavily focused on bioinformatics then you might not find any bioinformatics utilities available in the modules. For example the SUMMIT supercomputer (ALPINE’s predecessor) at Boulder had almost no bioinformatics modules. In such cases have to install your own software as described in Section 7.6.2.
The ALPINE supercomputer at Boulder now has some bioinformatics modules, which is handy.
In order to see them, you have to login to a compute node on ALPINE. There is much more
about that in the next chapter, but for now after logging in to login.rc.colorado.edu the
process looks like this:
# load a module that gives you slurm access to alpine
module load slurm/alpine
# get a shell on one of the interactive compute nodes on alpine
srun --partition atesting --pty /bin/bash
# once you get a new shell prompt after that, list available modules with:
module availThe bioinformatics modules listed this way, currently, on ALPINE are:
---------------------------------------------------------------------- Bioinformatics -----------------------------------------------------------------------
alphafold/2.2.0 bcftools/1.16 cellranger/7.1.0 homer/4.11 nextflow/23.04 (D) samtools/1.16.1
alphafold/2.3.1 (D) bedtools/2.29.1 cutadapt/4.2 htslib/1.16 picard/2.27.5 sra-toolkit/3.0.0
bamtools/2.5.2 bowtie2/2.5.0 fastqc/0.11.9 multiqc/1.14 plink2/2.00a2.3 star/2.7.10b
bbtools/39.01 bwa/0.7.17 gatk/4.3.0.0 nextflow/22.10.6 qiime2/2023.5 trimmomatic/0.39
While by no mean complete, this is a decent set of tools.
The module command has a large number of subcommands which are invoked
with a word immediately following the module command. We have already seen
how module avail lists the available modules. The other most important commands
appear in Table 7.3.
| Module Subcommand | What it does |
|---|---|
avail |
Lists available modules (i.e., software that can be loaded as a module) |
add modulefile |
same as load modulefile |
list |
list all currently loaded modulefiles. |
load modulefile |
add the necessary ingredients to one’s shell to be able to run the programs contained in modulefile. |
purge |
unload all the currently loaded modulefiles. |
rm modulefile |
same as unload modulefile. |
show modulefile |
describe the modulefile and the changes to the PATH and other shell evironment variables that occur when loading the modulefile |
unload modulefile |
reverse the changes made to
the shell environment made by
load modulefile |
Let’s play with the modulefiles (if any) on your HPCC! First, get an interactive session on a compute node. On SUMMIT:
Then list the modulefiles available:
You might notice that multiple versions of some programs are available, like:
R/3.3.0
R/3.4.3
R/3.5.0In such a case, the latest version is typically the default version that will be loaded when you request that a program modulefile be loaded, though you can specifically request that a particular version be loaded. On SUMMIT, check to make sure that the R program is not available by default:
You should be told:
bash: R: command not foundIf not, you may have already loaded the R modulefile, or you might have R available from
an activated conda environment (see below).
To make R available via module you use:
module load R
# now check to see if it works:
R
# check the version number when it launches
# to get out of R, type: quit() and hit RETURNTo list the active modulefiles, try:
This is quite interesting. It shows that several other modulefiles, above and beyond R, have been loaded as well. These are additional dependencies that the R module depends on.
To remove all the modulefiles that have been loaded, do:
If you are curious about what happens when a module file is loaded,
you can use the show subcommand, as in module show modulefile:
The output is quite informative.
To get a different version of a program available from module, just
include the modulefile with its version number as it appears when printed
from module avail, like:
module load R/3.3.0
# check to see which modulefiles got loaded:
module list
# aha! this version of R goes with a different version of the
# intel compiler...
# check the version number of R by running it:
R
# use quit() to get out of R.Once again, purge your modulefiles:
and then try to give the java command:
You should be told that the command java is not found.
There are several useful bioinformatics programs that are written in Java.
Java is a language that can run on many different computer architectures
without being specifically compiled for that architecture. However, in order to
run a program written in Java, the computer must have a “Java Runtime Environment” or JRE.
Running the java command above and having no luck shows that by default, SUMMIT (and most
supercomputers) do not have a JRE available by default. However, almost all HPCCs will
have a JRE available as in a modulefile containing the Java Development Kit, or JDK.
Look at the output of module avail and find jdk, then load that modulefile:
You will need to load the jdk module file in order to run the Java-based bioinformatics
program called GATK.
Note that every time you get a start a new shell on your HPCC, you will typically not have
any modulefiles loaded (or will only have a few default modulefiles loaded). For this reason
it is important, when you submit jobs using SLURM (see Section 8.4.2) that
require modulefiles, the module load modulefile command for those modules should appear
within the script submitted to SLURM.
Finally, we will note that the module utility works somewhat differently than the
conda environments described in the next section. Namely, conda environments
are “all-or-nothing” environments that include different programs. You can’t activate
a conda environment, and then add more programs to it by activating another
conda environment “on top of” the previous one. Rather, when activating a conda
environment, the configurations of any existing, activated environment are completely
discarded. By contrast, modulefiles can be layered on top of one another. So, for example,
if you needed R, samtools, and bcftools, and these were all maintained in
separate modulefiles on your HPCC, then you could load them all, alongside one another,
with:
Unlike a Miniconda environment, when you layer modulefiles no top of one another like this, conflicts between the dependencies may occur. When that happens, it is up to the sysadmins to figure it out. This is perhaps why the modulefiles on a typical HPCC may often carry older (if not completely antiquated) versions of software. In general, if you want to run newer versions of software on your HPCC, you will often have to install it yourself. Doing so has traditionally been difficult, but a packages management system called Miniconda has made it considerably easier today.
7.6.2 Miniconda
We will first walk you through a few steps with mamba to install some bioinformatic software into an environment on your cluster. After that we will discuss more about the underlying philosophy of Miniconda, and how it is operating.
7.6.2.1 Installing or updating mamba via miniforge
To do installation of software, you probably should not be on ALPINE’s login nodes. They offer “compile nodes” that should be suitable for installing with Miniconda. So, do this:
If you are on Hummingbird, be sure to get a
bashshell before doing anything else, by typingbash(if you have not already setbashas your default shell (see the previous chapter)).First, check if you have mamba or conda.
If you see some help information for
condathen you already have Miniconda (or Anaconda) and you could, if desired, update it with:Likewise, you could update mamba, if desired, with
If you have conda but not mamba, I would recommend removing your conda installation
and installing a fresh mamba installation. One may legitimately not want to do this if
they have lots of conda environments that they use for many different projects. That is the
way I used to use conda/mamba. Now, I tend to use mamba differently—only installing very
small environments in the course of my workflows, and I do feel a great deal of freedom knowing
that it is no big deal for me to completely erase mamba and all its environments and then reinstall it.
If you get an error telling you that your computer does not know about a command, conda or mamba
then you don’t have either and you can simply install mamba. Yay!
you do not have Miniconda and you must install it. You do that by downloading the Miniconda package
with wget and then running the Miniconda installer, like this:
```sh
# start in your home directory and do the following:
mkdir conda_install
cd conda_install/
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
chmod u+x Miniconda3-latest-Linux-x86_64.sh
./Miniconda3-latest-Linux-x86_64.sh
```That launches the Miniconda installer (it is a shell script). Follow the prompts and agree
to the license. Typically you would agree with the default install location; however,
the default install location is in the home directory, and you could quickly fill that up on
SUMMIT. So, set the install location to you projects directory. After that, be sure
to agree to initialize conda.
At the end, it tells you to log out of your shell and log back in for changes to take
effect. It turns out that it suffices to do cd ~; source .bash_profile
So, in summary, for SUMMIT users, after running the shell code in the above code block, you should:
- Press Enter to review the license agreement
- Hit the space bar to page through the agreement.
- At the end of the agreement, type yes to agree to it
- Next you are told where miniconda will be installed, but on ALPINE, you
do not want to install it in the default location in your home directory.
Instead, enter the location where you want it, namely,
/projects/your_csu_id@colostate.edu/miniconda3. Where you have changedyour_csu_idto be your CSU eID. When you type that in, you do not have to put a backslash before the@. - When asked if you wish the installer to run conda init, answer yes.
After that, you can logoff and log back on again, or, easier yet, you can just
type bash and that will initialize conda (by reading from your .bashrc which
the conda installer has modified). In the future, when you log in to a fresh shell
you should not have to type bash to get conda initialized.
Once you complete the above, your command prompt should have been changed to something that looks like:
The (base) is telling you that you are in Miniconda’s base environment. Typically you want to keep the
base environment clean of installed software, so we will almost always install software into a new environment.
At the end of this, you can cd back to your home directory and delete the ~/conda_install directory if you would like to.
7.6.2.2 Installing mamba
After a fresh install of conda, it is worth it to also install its faster,
fresher, younger cousin, mamba into your base environment. mamba is a tool
much like conda, and is a total replacement for it in some situations, and it is
recommended for installing Snakemake, which we will use later in the course. So,
if you have a new or freshly updated conda install, go ahead and do:
7.6.2.3 Installing software into a bioinformatics environment
If everything went according to plan above, then we are ready to use Miniconda to
install some software for bioinformatics. We will install a few programs that we will
use extensively in the next few weeks: bwa, samtools, and bcftools. We will
install these programs into a conda environment that we will name bioinf (short
for “bioinformatics”). It takes just a single command:
That should only take a few minutes, at most.
Note that if you installed mamba you could have done:
and gotten the same result. Just do it one way, though!
To test that we got the programs we must activate the bioinf environment, and then issue
the commands, bwa, samtools, and bcftools. Each of those should spit back some help
information. If so, that means they are installed correctly! It looks like this:
After that you should get a command prompt that starts with (bioinf), telling you that the
active conda environment is bioinf. Now, try these commands:
7.6.2.4 Uninstalling Miniconda and its associated environments
It may become necessary at some point to uninstall Miniconda. One important case of this is if you end up overflowing your home directory with conda-installed software. In this case, unless you have installed numerous, complex environments, the simplest thing to do is to “uninstall” Miniconda, reinstall it in a location with fewer hard-drive space constraints, and then simply recreate the environments you need, as you did originally.
This is actually quite germane to SUMMIT users. The size quota on home directories on SUMMIT is
only 2 Gb, so you can easily fill up your home directory by installing a few conda environments.
To check how much of the hard
drive space allocated to you is in use on SUMMIT, use the curc-quota command. (Check the documentation
for how to check space on other HPCCs, but note that Hummingbird users get 1 TB on their home
directories). Instead of
using your home directory to house your Miniconda software, on SUMMIT you can put it in your
projects storage area. Each user gets more storage (like 250 Gb) in a directory
called /projects/username where username is replaced by your SUMMIT username,
for example: /projects/eriq@colostate.edu
To “uninstall” Miniconda, you first must delete the miniconda3 directory in your
home directory (if that is where it got installed to). This can take a while. It is done with:
Then you have to delete the lines between # >>> and # <<<, wherever they occur in your ~/.bashrc and ~/bash_profile
files, i.e., you will have to remove all of the lines that look
something like thius:
# >>> conda initialize >>>
# !! Contents within this block are managed by 'conda init' !!
__conda_setup="$('/Users/eriq/miniconda3/bin/conda' 'shell.bash' 'hook' 2> /dev/null)"
if [ $? -eq 0 ]; then
eval "$__conda_setup"
else
if [ -f "/Users/eriq/miniconda3/etc/profile.d/conda.sh" ]; then
. "/Users/eriq/miniconda3/etc/profile.d/conda.sh"
else
export PATH="/Users/eriq/miniconda3/bin:$PATH"
fi
fi
unset __conda_setup
# <<< conda initialize <<<After all those conda lines are removed from your ~/.bashrc and ~/bash_profile, logging out
and logging back in, you should be free from conda and ready to reinstall it in
a different location.
To reinstall miniconda in a different location, just follow the
installation instructions above, but when you are running the
./Miniconda3-latest-Linux-x86_64.sh script, instead of choosing the default
install location, use a location in your project directory. For example, for me, that is:
/projects/eriq@colostate.edu/miniconda3.
Then, recreate the bioinf environment described above.
If you are having fun making environments and you think that you might like to use R on the cluster, then you might want to make an environment with some bioinformatics software that also has the latest version of R on miniconda installed. At the time of writing that was R 3.6.1. So, do:
That makes an environment called binfr (which turns out to also be way easier to type that bioinfr).
The r-essentials in the above command line is the name for a collection of 200 commonly used R packages (including
the tidyverse). This procedure takes a little while, but it is still far less painful than using the
version of R that is installed on SUMMIT with the modules packages, and then trying to build the tidyverse
from source with install.packages().
7.6.2.5 What is Miniconda doing?
This is a good question. We won’t go deeply into the specifics, but will skim the surface of a few topics that can help you understand what Miniconda is doing.
First, Miniconda is downloading programs and their dependencies into the miniconda3
directory. Based on the lists of dependencies and conflicts for each program that is being
installed, it makes a sort of “equation,” which it can “solve” to find the versions of
different programs and libraries that can be installed and which should “play nicely with
one another (and with your specific computer architecture.” While it is solving this
“equation” it is doing so while also doing its best
to optimize features of the programs (like using the latest versions, if possible).
Solving this “equation” is an example of a Boolean Satisfiability problem, which is a known
class of difficult (time-consuming) problems. If you are requesting a lot of programs, and
especially if you do not constrain your request (by demanding a certain version of
the program) then “solving” the request may take a long time. However, when installing
just a few bioinformatics programs it is unlikely to ever take too terribly long.
Once miniconda has decided on which versions of which programs and dependencies to install,
it downloads them and then places them into the requested environment (or the active environment
if no environment is specifically requested). If a program is installed into an environment, then you
can access that program by activating the environment (i.e. conda activate bioinf). Importantly,
if you don’t activate the environment, you won’t be able to use the programs installed there.
We will see later in writing bioinformatic scripts, you will always have to explicitly
activate a desired conda environment when you run a script on a compute node through the job
scheduler.
The way that Miniconda delivers programs in an environment is by storing all the programs
in a special environment directory (within the miniconda3/envs directory), and then, when
the environment is activated, the main thing that is happening is that conda is manipulating your
PATH variable to include directories within the environment’s directory
within the miniconda3/envs directory. An easy way to see this is simply by
inspecting your path variable while in different environments. Here we compare the PATH
variable in the base environment, versus in the bioinf environment, versus in the
binfr environment:
(base) [~]--% echo $PATH
/projects/eriq@colostate.edu/miniconda3/bin:/projects/eriq@colostate.edu/miniconda3/condabin:/usr/local/bin:/bin:/usr/bin:/usr/bin:/usr/local/sbin:/usr/sbin:/home/eriq@colostate.edu/bin:/home/eriq@colostate.edu/bin
(base) [~]--% conda activate bioinf
(bioinf) [~]--% echo $PATH
/projects/eriq@colostate.edu/miniconda3/envs/bioinf/bin:/projects/eriq@colostate.edu/miniconda3/condabin:/usr/local/bin:/bin:/usr/bin:/usr/bin:/usr/local/sbin:/usr/sbin:/home/eriq@colostate.edu/bin:/home/eriq@colostate.edu/bin
(bioinf) [~]--% conda activate binfr
(binfr) [~]--% echo $PATH
/projects/eriq@colostate.edu/miniconda3/envs/binfr/bin:/projects/eriq@colostate.edu/miniconda3/condabin:/usr/local/bin:/bin:/usr/bin:/usr/bin:/usr/local/sbin:/usr/sbin:/home/eriq@colostate.edu/bin:/home/eriq@colostate.edu/bin(To be sure, miniconda can change a few more things than just your PATH variable when you activate an environment, but
for the typical user, the changes to PATH are most important.)
7.6.2.6 What programs are available on Minconda?
There are quite a few programs for multiple platforms. If you are wondering
whether a particular program is available from Miniconda, the easiest first
step is to Google it. For example, search for miniconda bowtie.
You can also search from the command line using conda search. Note that most
bioinformatics programs you will be interested in are available on a conda
channel called bioconda. You probably saw the -c bioconda option
applied to the conda create commands above. That options tells conda to search
the Bioconda channel for programs and packages.
Here, you can try searching for a couple of packages that you might end up using to analyze genomic data:
7.6.2.7 Can I add more programs to an environment?
This is a worthwhile question. Imagine that we have been happily working in our bioinf conda environment
for a few months. We have finished all our tasks with bwa, samtools, and bcftools, but perhaps now we
want to analyze some of the data with angsd or plink. Can we add those programs to our
bioinf environment? The short answer is “Yes!”. The steps are easy.
Activate the environment you wish to add the programs to (i.e.
conda activate bioinffor example).Then use
conda install. For example to install specific versions ofplinkandangsdthat we saw above while searching for those packages we might do:
Now, the longer answer is “Yes, but…” The big “but” there occurs because if different
programs require the same dependencies, but rely on different versions of the dependencies,
installing programs over different commands can cause miniconda to not identify some
incompatibilities between program dependencies. A germane example occurs if you first install
samtools into an environment, and then, after that, you install bcftools, like this:
conda create -n samtools-first # create an empty environment
conda activate samtools-first # activate the environment
conda install -c bioconda samtools # install samtools
conda install -c bioconda bcftools # install bcftools
bcftools # try running bcftoolsWhen you try running the last line, bcftools barfs on you like so:
bcftools: error while loading shared libraries: libcrypto.so.1.0.0: cannot open shared object file: No such file or directorySo, often, installing extra programs does not create problems, but it can. If you find yourself battling
errors from conda-installed programs, see if you can correct that by creating a new environment and installing all the programs
you want at the same time, in one fell swoop, using conda create, as in:
7.6.2.8 Exporting environments
In our introduction to Miniconda, we mentioned that it is a great boon to reproducibility. Clearly, your analyses will be more reproducible if it is easier for others to install software to repeat your analyses. However, Miniconda takes that one step further, allowing you to generate a list of the specific versions of all software and dependencies in a conda environment. This list is a complete record of your environment, and, supplied to conda, it is a specification of exactly how to recreate that environment.
The process of creating such a list is called exporting the conda
environment. Here we demonstrate its use by exporting the bioinf
environment from SUMMIT to a simple text file. Then we use that text file
to recreate the environment on my laptop.
# on summit:
conda activate bioinf # activate the environment
conda env export # export the environmentThe last command above just sends the exported environment to stdout, looking like this:
name: bioinf
channels:
- bioconda
- defaults
dependencies:
- _libgcc_mutex=0.1=main
- bcftools=1.9=ha228f0b_4
- bwa=0.7.17=hed695b0_7
- bzip2=1.0.8=h7b6447c_0
- ca-certificates=2020.1.1=0
- curl=7.68.0=hbc83047_0
- htslib=1.9=ha228f0b_7
- krb5=1.17.1=h173b8e3_0
- libcurl=7.68.0=h20c2e04_0
- libdeflate=1.0=h14c3975_1
- libedit=3.1.20181209=hc058e9b_0
- libgcc-ng=9.1.0=hdf63c60_0
- libssh2=1.8.2=h1ba5d50_0
- libstdcxx-ng=9.1.0=hdf63c60_0
- ncurses=6.1=he6710b0_1
- openssl=1.1.1d=h7b6447c_4
- perl=5.26.2=h14c3975_0
- samtools=1.9=h10a08f8_12
- tk=8.6.8=hbc83047_0
- xz=5.2.4=h14c3975_4
- zlib=1.2.11=h7b6447c_3
prefix: /projects/eriq@colostate.edu/miniconda3/envs/bioinfThe format of this information is YAML (Yet Another Markup Language), (which we saw in the headers of RMarkdown documents, too).
If we stored that output in a file:
And then copied that file to another computer, then we can recreate the environment on that other computer with:
That should work fine if the new computer is of the same architecture (i.e., both
are Linux computers, or both are Macs). However, the specific build numbers
referenced in the YAML (i.e. things like the h7b6447c_3 part of the program name)
can create problems when installing on other architectures. In that case, we must
export without the build names:
Even that might fail if the dependencies differ on different architectures,
in which case you can export just the list of the actual programs that
you reqested be installed, by using the --from-history option. For example:
% conda env export --from-history
name: bioinf
channels:
- defaults
dependencies:
- bwa
- bcftools
- samtools
prefix: /projects/eriq@colostate.edu/miniconda3/envs/bioinfThough, even that fails, cuz it doesn’t list bioconda in there.
7.6.3 Installing Java Programs
Java programs run without compilation on many different computer architectures, so long as the computer has a Java Runtime Environment, or JRE. Thus, the steps to running a Java program are usually simply:
- Download the Java program, which is usually stored in what is called a Jar file, having a
.jarextension. - Ensure that a JRE is available (either loading a JRE or JDK modulefile, or, if that is not
available, creating a
condaenvironment that includes the JRE.) - Launch the Java program with
java -jar progpath.jarwhereprogpath.jaris the path to the Jar file that you have downloaded and wish to run.
7.6.3.1 Installing GATK
The most important Java based program for bioinformatics is GATK (and its companion
program, which is now a part of GATK, called PicardTools). This program,
since version 4,
comes with a python “wrapper” script that takes care of launching the GATK program
without using the java -jar syntax, and it also gives it a much more conventional
Unix command-line program “feel” than it had before, making it somewhat easier
to use if you are familiar with working on the shell.
Here, we describe how to download and install GATK for fairly typical or standard use
cases. There are further dependencies for some GATK analyses that can be installed
using Miniconda, but we won’t cover that here, as we don’t need those dependencies
for what we will be doing. (However, if you have digested the previous sections on Miniconda
you should have no problem installing the other dependencies with conda).
- Download the GATK package. GATK, since version 4, is available online
at GitHub using links found at https://github.com/broadinstitute/gatk/releases/tag/4.1.6.0. We use
wgeton the cluster to download this. I recommend creating a directory calledjava-programsfor storing your Java programs. If you are working on SUMMIT, this should go in yourprojectsdirectory, to avoid filling up your tiny home directory. At the time of writing, the latest GATK release was version 4.1.6.0. A later version may now be available, and the links below should be modified to get that later version if desired.
# replace user with your username
cd /projects/user\@colostate.edu/
mkdir java-programs # if you don't already have such a directory
cd java-programs # enter that directory
wget https://github.com/broadinstitute/gatk/releases/download/4.1.6.0/gatk-4.1.6.0.zip
# unzip that compressed file into a directory
unzip gatk-4.1.6.0.zip
# if that step was successful, remove the zip file
rm gatk-4.1.6.0.zip
# cd into the gatk directory
cd gatk-4.1.6.0
# finally, print the working directory to get the path
pwdWhen I do the last command, I get: /projects/eriq@colostate.edu/java-programs/gatk-4.1.6.0 You will
want to copy the path on your system so that you can include it in your ~/.bashrc file. In the following
I refer to the path to the GATK directory on your system as <PATH_TO>. You should replace <PATH_TO>
in the following with the paht to the GATK directory on your system. Edit your ~/.bashrc file,
adding the following lines above the >>> conda initialize >>> block:
On my system, it looks like the following when I have replaced
export PATH=$PATH:/projects/eriq@colostate.edu/java-programs/gatk-4.1.6.0
source /projects/eriq@colostate.edu/java-programs/gatk-4.1.6.0/gatk-completion.shOnce that is done, save and close ~/.bashrc and then source it for the changes to take
effect. (You don’t typically need to source it if you login to a new shell, but here,
since you are not opening a new shell, you need to source it.)
Now, you should be able to give the command
and you will get back a message about the syntax for using gatk. If not,
then something has gone wrong.
If gatk above worked as expected (gave you a help message), you are ready
to run a very quick experiment to test if we are all set
for calling variants (SNPs and indels) from the .bam files that were
created in the chr-32-bioinformatics homework. Be certain that you
are on a compute node before doing this. (Check it with hostname).
# cd to your homework folder. On my system, that is:
cd scratch/COURSE_STUFF/chr-32-bioinformatics-eriqande/
# make a file that holds the paths to all the duplicate-marked
# bam files you created during the homework:
ls -l mkdup/*_mkdup.bam | awk '{print $NF}' > bamfiles.list
# make sure bamfiles.list has the relative paths to a number
# of different bamfiles in it:
cat bamfiles.list
# make a directory to put the output into
mkdir vcf
# make sure the JRE is loaded (on SUMMIT)
module load jdk
# GATK needs two different indexes of the genome. Unfortunately
# the version we have is not compressed with bgzip, so
# we will have to make an uncompressed version of it and
# then index it. GATK expects the index (or, as they call it,
# the "dictionary" to be named a certain way...)
gunzip -c genome/GCA_002872995.1_Otsh_v1.0_genomic.fna.gz > genome/GCA_002872995.1_Otsh_v1.0_genomic.fna
conda activate bioinf
samtools faidx genome/GCA_002872995.1_Otsh_v1.0_genomic.fna
gatk CreateSequenceDictionary -R genome/GCA_002872995.1_Otsh_v1.0_genomic.fna \
-O genome/GCA_002872995.1_Otsh_v1.0_genomic.dict
# then we will launch GATK to do variant calling and create a VCF file
# from the BAMs in bamfiles.list in a 5 Kb region (we expect about
# 50 variants in such a small part of the genome) on Chromosome 32
# which is named CM009233.1
gatk --java-options "-Xmx4g" HaplotypeCaller \
-R genome/GCA_002872995.1_Otsh_v1.0_genomic.fna \
-I bamfiles.list \
-O vcf/tiny-test.vcf \
-L CM009233.1:2000000-2005000Once that finishes, look at the resulting VCF file:
more vcf/tiny-test.vcf
# if you get tired of scrolling through the header lines (with
# endless small genome fragment names). Then quit that (hit q)
# and view it without the header:
bcftools view -H vcf/tiny-test.vcfIf that looks like a bunch of gibberish, rejoice! We will learn about the VCF file format soon!
7.7 vim: it’s time to get serious with text editing
Introduce newbs to the vimtutor.
Note, on a Mac, add these lines to your ~/.vimrc file:
filetype plugin indent on
syntax onThat will provide syntax highlighting.
7.7.1 Using neovim and Nvim-R and tmux to use R well on the cluster
These are currently just notes to myself. And I won’t end up doing this anyway.. I should probably replace with with my own rView package…
On Summit you can follow the directions install Neovim and Nvim-R etc, found at section 2 of https://gist.github.com/tgirke/7a7c197b443243937f68c422e5471899#ucrhpcc. You can just do 2.1 to 2.6. 2.7 is the routine for user accounts. You don’t need to install Tmux.
You need to get an interactive session on a compute node and then
The last two are needed to get a random number to start up client through R. It is amazing to me that they call a specific Intel library to do that, and apparently loading the R module alone doesn’t get you that.
Uncomment the lines:
let R_in_buffer = 0
let R_tmux_split = 1 in your ~/.config/nvim/init.vim. Wait! You don’t want to do that, necessarily, because tmux with NVim-R
is no longer supported (Neovim now has native terminal splitting support.)