Chapter 16 DNA Sequences and Sequencing
To understand the fundamentals of alignment of DNA sequence to a reference genome, and all the intricacies that such a process entails, it is important to know a few important facts about DNA and how the sequence of a DNA molecule can be determined. This section may be review for some, but it presents the minimal set of knowledge needed to understand how DNA sequencing works, today.
16.1 DNA Stuff
In bioinformatics, we are primarily going to be interested in representing DNA molecules in text format as a series of nucleotide bases (A, C, G, T). For the most part, we don’t want to stray from that very simple representation; however, it is important to understand a handful of things about DNA directionality and the action of DNA polymerase during the process of DNA replication to understand next generation sequencing and DNA alignment conventions.
DNA typically occurs as a double-helix of two complementary strands. Each strand is composed of a backbone of phosphates and deoxyribose molecules, to which DNA bases are attached. Figure 16.1 shows this for a fragment of double-stranded DNA of four nucleotides.
FIGURE 16.1: Schematic of the structure of DNA. (Figure By Madprime (talk—contribs) CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=1848174)
The orange parts show the ribose molecules in the backbone, while the four remaining colors denote the nucleotide bases of adenine (A), cytosine (C), guanine (G), and thymine (T). Each base on one strand is associated with its complement on the other strand. The hydrogen bonds between complements is what holds the two strands of DNA together. A and T are complements, and C and G are complements. (I remember this by noting that C and G and curvy and A and T are sharp and angular, so the pairs go together…).
Immediately this raises two challenges that must be resolved for making a simple, text-based representation of DNA:
- When describing a sequence of DNA bases, which direction will we read it in?
- Which strand will we read off of?
We leave the second question until later. But note that the order in which DNA sequence is read is, by convention, from the 5’ to the 3’ end of the molecule. The terms 3’ and 5’ refer to different carbon atoms on the ribose backbone molecules. In the figure above, each little “kink” in the ribose molecule ring (and in the attached lines) is a carbon molecule. If you count clockwise from the oxygen in ribose, you see that the third carbon (the 3’ carbon) is the carbon atom that leads to a phosphate group, and through the phosphate, to an attachment with the 5’ carbon atom of the next ribose. The 3’ and 5’ carbon atoms are labelled in red on the top-left ribose molecule in Figure 16.1.
Notice that when we speak of reading a DNA sequence, we are implicitly talking about reading the sequence of one of the two strands of the double-stranded DNA molecule. I’ll say it again: a DNA sequence is always the sequence of one strand of the molecule. But, if that DNA were “out living in the wild” it would have been in double-stranded form, and would have been paired with its complement sequence. For now, just note that any DNA sequence always has a complement which is read in the reverse direction. Thus, if we have a sequence like:
5'--ACTCGACCT--3'Then, paired with its complement it would look like:
5'--ACTCGACCT--3'
|||||||||
3'--TGAGCTGGA--5'and, if you were to write its complement in standard 5’ to 3’ order, you would have to reverse it like so:
5'--AGGTCGAGT--3'16.1.1 DNA Replication with DNA Polymerase
So, why do we read DNA sequence from 5’ to 3’? Is it just because geneticists are wacky, backwards folks and thought it would be fun to read in a direction that sounds, numerically, to be backwards? No! It is because \(5'\longrightarrow 3'\) is the direction in which a new strand of DNA is synthesized during DNA replication.
When Watson and Crick (1953) published the first account of the double helical structure of DNA, they noted that the double-helix (i.e., two-stranded) nature of the molecule immediately suggested a copying mechanism (Figure 16.2).
FIGURE 16.2: Excerpt from Crick and Watson (1953).
(I’ve also included, in the excerpt, the inadequate acknowledgment of the centrality of Rosalind Franklin’s pioneering X-ray crystallography results to Crick and Watson’s conclusions—an issue in scientific history about which much has been written, see, for example, this article in The Guardian.)
Figure 16.3 shows a schematic of what DNA looks like during the replication process.
FIGURE 16.3: DNA during replication. (Figure adapted from the one by Madprime (talk—contribs) CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=1848174)
Essentially, during DNA replication, a DNA polymerase molecule finds nucleotide bases (attached to three phoshpate groups) to build a new strand that is complementary to the DNA template strand, and it guides those nucleotide triphosphates to the appropriate place in the complementary strand and helps them be incorporated into that growing, complementary strand. The newly syntesized strand is a reverse complement of the template strand. However, DNA polymerase is not capable of “setting up shop” anywhere upon a template strand and simply stuffing complementary bases in wherever it wants. In fact, DNA polymerase is only able to add new bases to a growing strand if it can attach the new nucleotide triphosphate to a free 3’ hydroxyl that is on the end of the growing strand (the 3’ hydroxyl is just a hydroxyl group attached to the 3’ carbon). In Figure 16.3, the template strand is the strand on the right of the figure, and the growing complementary strand is on the left side. There is a free 3’ hydroxyl group on the ribose attached to the cytosine base. That is what is needed for DNA polymerase to be able to place a thymine triphosphate (complementary to adenine on the template strand) in the currently vacant position. If that thymine comes with a free 3’ hydroxyl group, then DNA polymerase will next place a guanine (complementary to the cytosine on the template strand) on the growing chain. And so forth. Thus, we see how the new strand of DNA is synthesized from the 5’ to the 3’ end of the growing chain.
Of course, some people find it easier to think about a new strand of DNA being sythesized in the 3’ to 5’ direction along the template strand. This is equivalent. However, if you just remember that “free three” rhymes, and that DNA polymerase needs a free 3’ hydroxyl to add a new base to the growing strand, you can always deduce that DNA must “grow” in a 5’ to 3’ direction.
16.1.2 The importance of the 3’ hydroxyl…
It would be hard to overstate the importance to molecular biology of DNA polymerase’s dependence upon a free 3’ hydroxyl group for new strand synthesis. This simple fact plays a central role in:
- polymerase chain reaction (PCR)—the PCR primers are little oligonucleotides that attach to a template strand and provide a free 3’ hydroxyl group for the initiation of synthesis.
- a ddNTP is a nucleotide attached to a ribose molecule that lacks a hydroxyl group on its 3’ carbon. Incorporation of such a ddNTP into a growing DNA strand terminates further DNA extension, and forms the basis for Sanger sequencing (we’ll explore this below).
- Some medications are designed to interfere with viral DNA replication. For example, AZT, or azino-thymine, is an anti-retroviral drug used to slow the progression of AIDS. It is a thymine nucleotide with an azino (\(\mathrm{N}_3\)) group (instead of a hydroxyl group) attached to the 3’ carbon. Azino-thymine is used preferentially by reverse transcriptase when synthesizing DNA. Incorporation of it into a growing chain terminates DNA synthesis.
The reversible inhibition of DNA extension also plays an important role in sequencing by synthesis as used by Illumina platforms. We will discuss this in a moment, but first we take a stroll down memory lane to refresh our understanding of Sanger sequencing so as to understand how radically different next-generation sequencing technologies are.
16.2 Sanger sequencing
It is hard to imagine that the first public human genome was sequenced almost entirely by Sanger sequencing. We discuss the Sanger sequencing method here so we can contrast it with what happens on, say, an Illumina machine today.
To perform Sanger sequencing, first it was necessary to do PCR to create numerous copies of a double stranded DNA fragment that was to be sequenced. For example let’s say that one wanted to sequence the 20-mer shown below, represented as double stranded DNA.
5'--AGGCTCAAGCTTCGACCGT--3'
3'--TCCGAGTTCGAAGCTGGCA--5'For Sanger sequencing, first, one would do PCR to create bazillions of copies of that double-stranded DNA. Then four separate further PCR reactions would be done, each one having been “spiked” with a little bit of one of four different ddNTPs which, if incorporated into the growing strand allow no further extension of it.
For example, if PCR were done as usual, but with the addition of ddATP, then occasionally, when a ddATP (an A lacking a 3’ hydroxyl group) is incorporated into the growing strand that strand will grow no more. Consequently, the products of that PCR (incorporating an appropriate concentration of ddATPs), once filtered to retain only the top strand from above, will include the fragments
## [1] "A*" "AGGCTCA*"
## [3] "AGGCTCAA*" "AGGCTCAAGCTTCGA*"Where the * follows the sequence-terminating base.
Likewise, in a separate reaction, occasional incorporation of a ddCTP will yield products:
## [1] "AGGC*" "AGGCTC*"
## [3] "AGGCTCAAGC*" "AGGCTCAAGCTTC*"
## [5] "AGGCTCAAGCTTCGAC*" "AGGCTCAAGCTTCGACC*"And in another reaction, occasional incorporation of ddGTP yields:
## [1] "AG*" "AGG*"
## [3] "AGGCTCAAG*" "AGGCTCAAGCTTCG*"
## [5] "AGGCTCAAGCTTCGACCG*"And in a final, separate reaction, incorporation of ddTTP would give:
## [1] "AGGCT*" "AGGCTCAAGCT*"
## [3] "AGGCTCAAGCTT*" "AGGCTCAAGCTTCGACCGT*"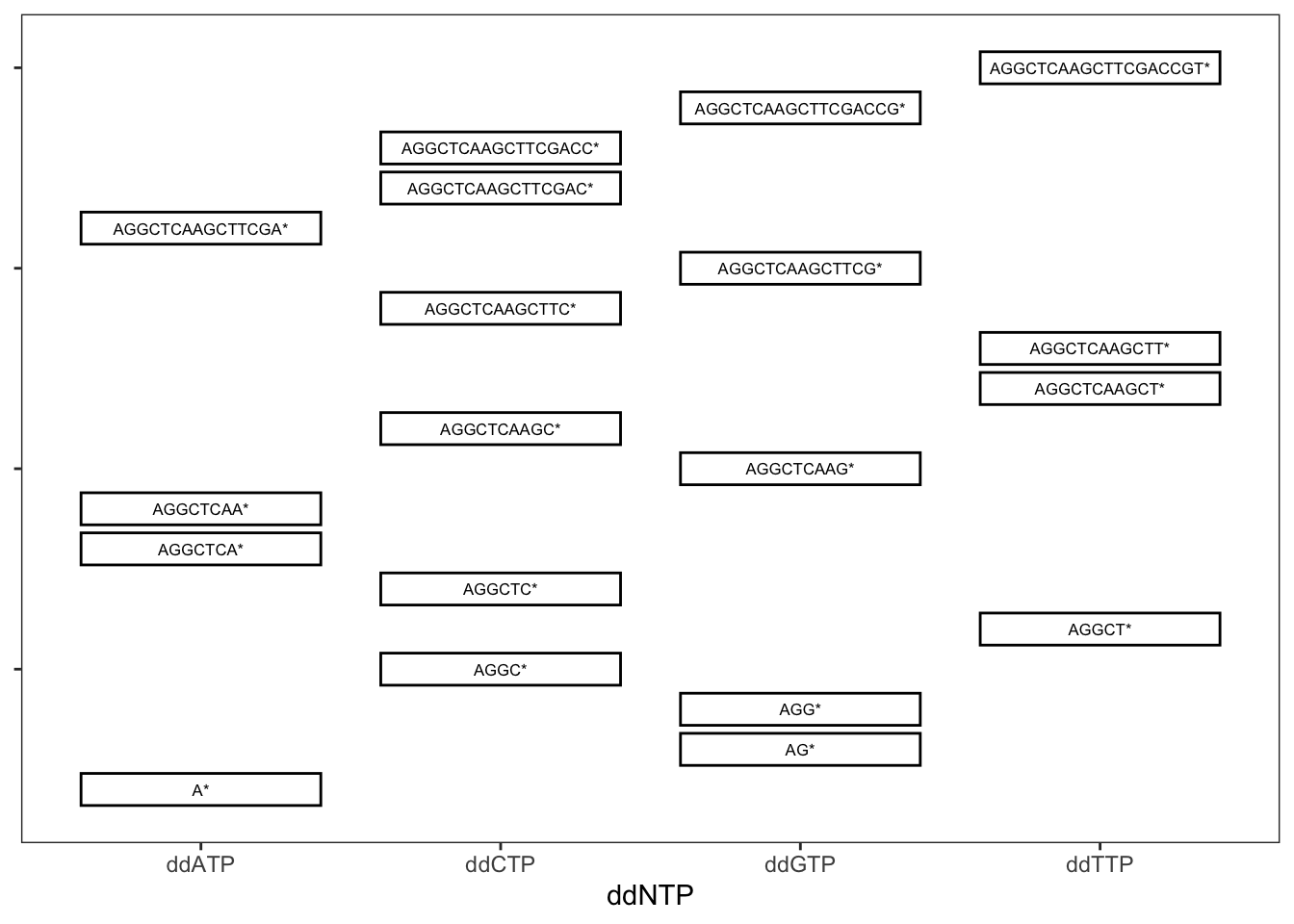
FIGURE 16.4: Stylized gel from Sanger sequencing. DNA migrates downward from the top, with smaller fragments traveling further than longer fragments. The four differnent lanes corresponds to four different PCR reactions incorporating ddNTPs of different types, as denoted below the x axis.

FIGURE 16.5: Stylized gel from Sanger sequencing. Here, the DNA sequence of each band cannot be seen.
However, even with the DNA fragemnt sequences obscured, their sequences can be detemined by working from the bottom to the top and adding a different DNA base according to which column the band is in. Try it out.
Some very important points must be made about Sanger sequencing:
- The signal obtained from sequencing is, in a sense, a mixture of the starting templates. So, if you have DNA from an individual, you have two copies of each chromosome, and they might carry slightly different sequences. At heterozygous sites, it is impossible to tell which allele came from which chromosome.
- To conduct this procedure, it was typical that specific PCR primers were used to sequence only a single fragment of interest. Extending the sequencing beyond that region often involved a laborious process of “walking” primers further out in either direction. Very tedious.
- Each sequencing reaction was typically carried out for just a single individual at once.
- Until a little over a decade ago, this is how sequencing was done in conservation genetics.
16.3 Illumina Sequencing by Synthesis
Illumina paired-end sequencing is currently the leading technology in conservation genomics.
They say that a picture is worth a thousand words, so a video may well be worth ten thousand. Illumina has a very informative video about sequencing by synthesis.
I have used some code (I found on GitHub) to provide captions to the video. These captions
include comments, as well as questions that form part of this week’s homework. (Yay!)
You can see the video at
https://eriqande.github.io/erics-captioned-vids/vids/illumina-sbs/
The main take-home messages I want everyone to get about Illumin sequencing is:
- The signal obtained from each cluster is the sequence of a single, single-stranded DNA fragment
- In paired-end sequencing, sequence from both ends of the fragment (but not necessarily the middle) is obtained.
- The technique lends itself to sequencing millions of “anonymous” chunks of DNA.
- The indexes, or “barcodes” allow DNA from multiple individuals to be sequenced in a single run.
- This is how most high-throughput sequencing is done today.