library(gscramble)
library(tidyverse)
#> ── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
#> ✔ dplyr 1.2.1 ✔ readr 2.2.0
#> ✔ forcats 1.0.1 ✔ stringr 1.6.0
#> ✔ ggplot2 4.0.3 ✔ tibble 3.3.1
#> ✔ lubridate 1.9.5 ✔ tidyr 1.3.2
#> ✔ purrr 1.2.2
#> ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
#> ✖ dplyr::filter() masks stats::filter()
#> ✖ dplyr::lag() masks stats::lag()
#> ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errorsInput Data
‘gscramble’ operates only for diploid species. The main functions in
gscramble require that genetic data be provided in R
objects (matrices/tibbles/etc.) with particular formatting. We describe
the formats of those objects here. Later, in the section, To and From plink Format, we
describe functions for reading data from PLINK (.ped / .map) format (and
perhaps others, in the future) into these necessary objects for
‘gscramble’.
The main required objects (loosely aligning with .bed/.fam/.bim file structure of binary PLINK files) are:
- A matrix of genotype data (the
Genoobject included with the package as example data) - A tibble of meta data about the individuals in the genotype data
(
I_metaas example data) - A tibble of meta data about the markers in the genotype data
(
M_metaas example data)
In addition to these objects, if you want to simulate hybrid individuals according to a pedigree, you will need to provide:
- A tibble that describes the rates of genetic recombination along the
chromosomes (
RecRates) - A tibble that describes a pedigree of interest within which sections
of chromosomes are segregated down successive generations, which we call
a “genome simulation pedigree” (
GSP) - A tibble that translates the population specifiers in the pedigree
(i.e.,
GSP) with the population/group labels associated with the genetic data (i.e.,I_meta) for different hybridization replicates (RepPop)
Examples of these objects (which have been heavily downsampled to
make them small enough to host on CRAN) illustrating 78 individuals
genotyped at 100 loci distributed across 3 chromosomes (chr 12, 17,
& 18) and sampled from 4 groups (Pop1, 2, 3, 4) from a data set of
invasive feral swine, are included as data objects with the package
(Geno, I_meta, and M_meta). The
full data set, from which the package data were downsampled, was used in
Smyser et al. (2020), and is available at https://datadryad.org/dataset/doi:10.5061/dryad.jsxksn05z.
Matrix of genotype data. Geno
The genotype data must be provided in a matrix. If there are
individuals and
loci in the diploid species, this matrix has
rows and
columns. Each locus gets two adjacent columns (one allele in each
column) and each individual gets one row. For example, in the first row
and first column Geno[1,1] is the first allele for the
first locus for the first individual. In the first row and second column
Geno[1,2] is the second allele for the first locus for the
first individual. In the first row and third column
Geno[1,3] is the first allele at the second locus for the
first individual, and so on.
The names/IDs of the individuals must be included as the the rownames of the matrix of genotypes.
Here is an example of the first 6 loci for the first 4 individuals
from the data included with the package as Geno:
Geno[1:4, 1:12]
#> [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11] [,12]
#> ID0033692 "G" "G" NA NA "T" "T" "A" "A" "G" "G" "T" "T"
#> ID0014942 "G" "G" "C" "C" "T" "T" "A" "A" "G" "G" "T" "T"
#> ID0014961 "G" "G" "C" "C" "T" "T" "A" "A" "G" "G" "T" "T"
#> ID0016971 "G" "G" "C" "C" "T" "T" NA NA "G" "G" "T" "T"These data must be stored as a character Matrix.
Don’t pass in a matrix of integers. The alleles can be any characters
whatsoever. This allows the data to be microsatellites,
("112", "116", etc), or microhaplotypes
("ACCGA", "ACCTC", etc.), or SNPs
("A", "C", "G",
"T"), etc. If you do have a matrix of integers, named, for
example IntMat, you can coerce all the elements of that
matrix to be characters without losing the matrix shape of the data, by
doing this:
storage.mode(IntMat) <- "character"Missing genotype data must be denoted by the standard R missing-data
specifier, NA. Don’t specify missing data as
"-1" and expect it to work properly! Change those
"-1"’s to NAs, or they will be regarded as an
allelic type, rather than as missing data. If you do have missing data
that are denoted as "-1", for example, then you can change
all those "-1"’s to NAs using the
following:
Geno[Geno == "-1"] <- NAPresently, since there are no "-1" entries in the
Geno dataset, this line changes nothing.
Individual meta data. I_meta
This is a tibble that gives information about the individuals whose
genotypes are in Geno. This can have as many different
columns as you want, but there must be at least two columns:
-
group: a column that gives the character name of the group/cluster/population that each individual is considered to be a part of. -
indiv: a column that gives the character ID of each individual.
The number of rows of this tibble should be exactly equal to the
number of rows on Geno and the order of
individuals in I_meta must correspond exactly to the order
of individuals in Geno.
Here is what the first few rows from the example data
I_meta look like:
head(I_meta)
#> # A tibble: 6 × 2
#> group indiv
#> <chr> <chr>
#> 1 Pop1 ID0033692
#> 2 Pop1 ID0014942
#> 3 Pop1 ID0014961
#> 4 Pop1 ID0016971
#> 5 Pop1 ID0016972
#> 6 Pop1 ID0017013Marker meta data. M_meta
This input is a tibble of information about the markers in the
Geno matrix. It can have a variety of columns in it, but it
is required to have three:
-
chrom: the character name of the chromosome upon which the marker occurs. For example,"1","X"or"Omy28", or as illustrated inM_meta:"chr12","chr17", and"chr18".
Importantly, if you are simulating physical linkage with recombination, then the names of the chromosomes in this file must correspond exactly to the names of the chromosomes inRecRates(see next section). -
pos: a numeric (integer or double) column giving the position of the marker (typically in base pairs, but it could be in arbitrary units that correspond to position units inRecRates), along the chromosome. These position values must be greater than 0. -
variant_id: a character vector of unique ID names for the markers. These should be globally unique, i.e., there CANNOT be two or more markers, even on different chromosomes, that are named the same thing.
There must be exactly half as many rows in M_meta as
there are columns in Geno, and the order of markers in
M_meta must correspond exactly to the order of markers in
the columns in Geno.
Here are the first few rows of the example data
M_meta:
head(M_meta)
#> # A tibble: 6 × 3
#> chrom pos variant_id
#> <chr> <dbl> <chr>
#> 1 12 4469057 WU_10.2_12_4469057
#> 2 12 5238225 ALGA0064411
#> 3 12 7394362 WU_10.2_12_7394362
#> 4 12 7651064 ASGA0090707
#> 5 12 8971475 WU_10.2_12_8971475
#> 6 12 11034660 WU_10.2_12_11034660Recombination rates. RecRates
This is a tibble that gives information about the rate of
recombinations in the genome. This is necessary if simulating linked
markers. It is not required in ‘gscramble’ to know a crossover rate nor
a recombination rate between every adjacent pair of markers (though if
you have that information, you can provide it in RecRates,
see below). Rather, the rate of recombination can be specified in terms
of the per-meiosis probability of recombination in a number of
(preferably relatively short—for example, one megabase or less) bins.
RecRates is a tibble which is required to have four
columns:
-
chrom: the chromosome on which the bin occurs. Note that the chromosome nomenclature must match exactly that used inM_meta. -
chrom_len: the length of the chromosome. Yes, the value for each chromosome will be the same for each marker/row associated with a givenchrom. -
start_pos: the starting position of the bin. This will typically be a position along the chromosome in base pairs, though this position can be in some other units, so long as it corresponds to the position used inM_meta. -
end_pos: the ending position of the bin. -
rec_prob: the per-meiosis probability of a recombination occurring in the bin.
There are some important notes:
- The start point of a bin should be 1 greater than the end point of the preceding bin.
- The positions of all the markers (in
M_meta) should be included amongst the bin intervals defined bystart_posandend_pos. Most crucially in this regard, the smalleststart_posshould be less than the smallestposinM_metaand the greatestend_posshould be greater than or equal to the largestposinM_meta. To enforce the latter, it is required that theend_posof the rightmost bin on each chromosome must be 1 greater than the chromosome length. Otherwise, recombination might never be possible between some pairs of markers in the data set. - The chromosome length in
chrom_lenmust exceed the position of every marker on the chromosome inM_meta. If this is not the case then it would be possible that some markers would be dropped from the data set, possibly with unexpected or bad results. You can check for errors in this regard by making sure that you pass your marker meta data (M_metain this example) to theMMoption of thesegregate()function (see below).
head(RecRates)
#> # A tibble: 6 × 5
#> chrom chrom_len start_pos end_pos rec_prob
#> <chr> <dbl> <dbl> <dbl> <dbl>
#> 1 12 63582536 1 79501 0.000694
#> 2 12 63582536 79502 1573908 0.00131
#> 3 12 63582536 1573909 2097503 0.000554
#> 4 12 63582536 2097504 3308663 0.00557
#> 5 12 63582536 3308664 4021031 0.0179
#> 6 12 63582536 4021032 5026780 0.0115Making a RecRates-like tibble from a plink map
file
The package includes a function plink_map2rec_rates()
that will turn a plink map file into a tibble formatted like
RecRates. Read the documentation for it with
?plink_map2rec_rates.
Genome Simulation Pedigree. GSP
The GSP is a specification of a pedigree within which
sections of chromosome will get segregated across the successive
generations represented within the pedigree and is used to to guide
sampling of these chromosomal sections without replacement. This will
allow us to characterize/simulate hybrid individuals for the purpose of
assessing the power to classify individuals to hybrid classes of
interest (i.e., F1s, F1BC1s, F2BC1s, etc.).
The GSP must be a tibble in which each individual has a
numeric identifier (from 1 up to the number of individuals in the
pedigree). Founders are listed directly (1…nFounders) whereas numbers
can represent more than one individual in successive generations (e.g.,
two F1 individuals sampled within the most basic pedigree illustrated
below are characterized as ‘3’). The founders’ parents are listed as NA
whereas non-founders have parents listed. Founders’ haplotypes must have
unique IDs and must originate from a specified population (typically
given in capital letters.) Individuals can be sampled from individuals
in the pedigree, and it is up to the user to indicate how many gametes
must be segregated to each individual (from each of its two parents) in
order to consume all the genetic material present among the
founders.
This probably sounds a little abstract, and, indeed, it is. To
demonstrate, ‘gscramble’ includes 15 basic GSPs (illustrated within the
object GSP_opts) that can be retrieved by specifying the
types of individuals of interest with the function
create_GSP(). As we will illustrate below, you are not
restricted to these GSP configurations. These 15 specific GSP
configurations were of interest to the authors and are included to
illustrate how to structure GSP tibbles that may be of interest to
you.
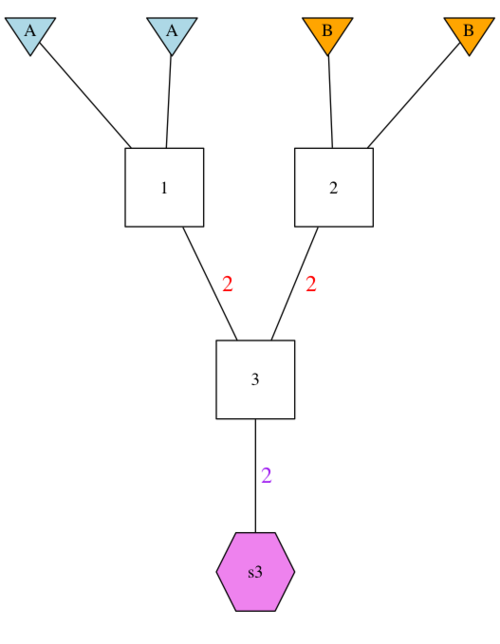
A Simple GSP. Here we have a pedigree with diploid founder ‘1’, who is formed by the union of two gametes from Population A, and diploid founder ‘2’ with two gametes from Population B. Founder ‘1’ and founder ‘2’ then pass 2 gametes (red 2’s) for the formation of 2 individuals represented by their descendant ‘box 3’. The pink hexagon ‘s3’ then represents the samples taken from ‘box 3’ with the pink 2 used to indicate the number of diploid individuals sampled.
Let’s develop this basic example of simulating F1 hybrids. The first
step would be to define the GSP in which we are describing sources as
‘p1’ and ‘p2’, and then we will need an associated RepPop
to represent the two different populations the founders belong to.
gspF1 <- create_GSP(pop1 = "p1", pop2 = "p2", F1 = TRUE)
gspF1
#> # A tibble: 3 × 11
#> ind par1 par2 ipar1 ipar2 hap1 hap2 hpop1 hpop2 sample osample
#> <dbl> <dbl> <dbl> <dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <dbl>
#> 1 1 NA NA NA NA 1a 1b p1 p1 NA NA
#> 2 2 NA NA NA NA 2a 2b p2 p2 NA NA
#> 3 3 1 2 2 2 NA NA NA NA s3 2To accompany this GSP, we next need to define the groups that we are
interested in hybridizing - in this case Pop1 and Pop2 - with a
RepPop tibble.
We will fully describe RepPop tibbles below. However,
GSPs need to be accompanied by a RepPop tibble that defines
the groups to be hybridized. Accordingly, let’s lightly introduce the
RepPop object here by specifying the tibble that
accompanies this F1 GSP.
Pattern = c("Pop1", "Pop2")
RepPopSimpleF1 <- tibble(
index = rep(1:1, each = 2),
pop = rep(c("p1", "p2"), times = 1),
group = Pattern
)
RepPopSimpleF1
#> # A tibble: 2 × 3
#> index pop group
#> <int> <chr> <chr>
#> 1 1 p1 Pop1
#> 2 1 p2 Pop2Please note that F1s are symmetrical, such that a Pop1-Pop2 F1 hybrid is identical to a Pop2-Pop1 F1 hybrid. This will not be the case as we continue developing more complex GSPs that include backcrossed hybrids.
This simplistic example specifies F1 hybrids between Pop1 and Pop2.
If we want to
add a little more complexity working within the same GSP, we could
modify the RepPop tibble to simulate hybrids between Pop1
and all other populations.
Pattern = c("Pop1", "Pop2", "Pop1",
"Pop3", "Pop1", "Pop4")
RepPopSimpleF1_b <- tibble(
index = rep(1:3, each = 2),
pop = rep(c("p1", "p2"), times = 3),
group = Pattern
)
RepPopSimpleF1_b
#> # A tibble: 6 × 3
#> index pop group
#> <int> <chr> <chr>
#> 1 1 p1 Pop1
#> 2 1 p2 Pop2
#> 3 2 p1 Pop1
#> 4 2 p2 Pop3
#> 5 3 p1 Pop1
#> 6 3 p2 Pop4Or, perhaps, let’s say you are interested in simulating F1 hybrids
for all pairwise combinations of populations (with the example data
Pop1, Pop2, Pop3, Pop4).
Accordingly, we need to specify the RepPop tibble to define
which populations to use in each replicate of the simulation:
Pattern = c("Pop1", "Pop2", "Pop1", "Pop3", "Pop1", "Pop4",
"Pop2", "Pop3", "Pop2", "Pop4", "Pop3", "Pop4")
RepPopSimpleF1_c <- tibble(
index = rep(1:6, each = 2),
pop = rep(c("p1", "p2"), times = 6),
group = Pattern
)
head(RepPopSimpleF1_c)
#> # A tibble: 6 × 3
#> index pop group
#> <int> <chr> <chr>
#> 1 1 p1 Pop1
#> 2 1 p2 Pop2
#> 3 2 p1 Pop1
#> 4 2 p2 Pop3
#> 5 3 p1 Pop1
#> 6 3 p2 Pop4Building upon this simplistic GSP, let’s define something a little
more ‘complex’ with F1s, F1BC1s, and F1BC2s.
Start by using the built-in function create_GSP().
gspComplex <- create_GSP(
pop1 = "p1",
pop2 = "p2",
F1 = TRUE,
F1B = TRUE,
F1B2 = TRUE
)
gspComplex
#> # A tibble: 7 × 11
#> ind par1 par2 ipar1 ipar2 hap1 hap2 hpop1 hpop2 sample osample
#> <dbl> <dbl> <dbl> <dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <dbl>
#> 1 1 NA NA NA NA 1a 1b p1 p1 NA NA
#> 2 2 NA NA NA NA 2a 2b p1 p1 NA NA
#> 3 3 NA NA NA NA 3a 3b p1 p1 NA NA
#> 4 4 NA NA NA NA 4a 4b p2 p2 NA NA
#> 5 5 1 4 2 2 NA NA NA NA s5 1
#> 6 6 2 5 2 2 NA NA NA NA s6 1
#> 7 7 3 6 2 2 NA NA NA NA s7 2For this example, Pop1 is our population of interest. We are simulating Pop1-Pop2 F1s, then pairing those F1 individuals with Pop1 to generate backcrossed hybrids. Note that these simulated F1B1s and F1BC2 are different than hybrids formed from Pop1-Pop2 F1s that are backcrossed to Pop2. Here is a picture of what this example would look like:

If we are interested in F1 hybrids between Pop 1 and each of the
other populations (Pop2, Pop3, or Pop4), as well as two generations of
backcrossing to each of those populations, we can use a
RepPop tibble that looks like this:
Pattern = c("Pop1", "Pop2", "Pop1",
"Pop3", "Pop1", "Pop4")
RepPopComplex1 <- tibble(
index = rep(1:3, each = 2),
pop = rep(c("p1", "p2"), times = 3),
group = Pattern
)
RepPopComplex1
#> # A tibble: 6 × 3
#> index pop group
#> <int> <chr> <chr>
#> 1 1 p1 Pop1
#> 2 1 p2 Pop2
#> 3 2 p1 Pop1
#> 4 2 p2 Pop3
#> 5 3 p1 Pop1
#> 6 3 p2 Pop4We will go over more examples of how to define populations in
RepPop in the section “Mapping populations/collections to
founding populations. RepPop” of this tutorial. But for
now, let’s examine GSPs further.
In our previous examples, we specified an initial suite of GSPs that
were of interest, combining various configurations of F1s, F2s, F1BC1s,
and F1BC2s using the create_GSP() function. However, you
are not limited to these GSPs and may create your own.
Here are two illustrations of more complex patterns of hybridization:
The package data object GSP shows an example of a genome
simulation pedigree with 13 members. Here is a picture of what it looks
like:

Note, you can produce this type of plot using the
gsp2dot() function in the ‘gscramble’ package, as follows,
but it requires the installation of the GraphViz dot
software.
csv <- system.file("extdata/13-member-ped.csv", package = "gscramble")
gsp_tib <- readr::read_csv(csv)
paths <- gsp2dot(g = gsp_tib, path = "images/13-member-ped")
# now, get rid of the dot and png files
file.remove(paths[1:2])The tibble specification of that same GSP is printed here:
GSP
#> # A tibble: 13 × 11
#> ind par1 par2 ipar1 ipar2 hap1 hap2 hpop1 hpop2 sample osample
#> <dbl> <dbl> <dbl> <dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <dbl>
#> 1 1 NA NA NA NA 1a 1b A A NA NA
#> 2 2 NA NA NA NA 2a 2b A A NA NA
#> 3 3 NA NA NA NA 3a 3b B B NA NA
#> 4 4 NA NA NA NA 4a 4b A A NA NA
#> 5 5 NA NA NA NA 5a 5b B B NA NA
#> 6 6 NA NA NA NA 6a 6b B B NA NA
#> 7 7 2 3 2 2 NA NA NA NA s7 1
#> 8 8 4 5 2 2 NA NA NA NA s8 1
#> 9 9 1 7 1 1 NA NA NA NA NA NA
#> 10 10 7 8 1 1 NA NA NA NA s10 1
#> 11 11 8 6 1 1 NA NA NA NA NA NA
#> 12 12 9 11 2 2 NA NA NA NA s12 2
#> 13 13 1 6 1 1 NA NA NA NA s13 1and, a CSV file with the same table in it can be found using:
system.file("extdata/13-member-ped.csv", package = "gscramble")A four-population GSP. gsp4
Or, alternatively, let’s say you are interested in patterns of hybridization that are not restricted to simply two populations.
For illustration, we will have another pedigree that represents an F1
between populations A and B then mating with an F1 from populations C
and D. The tibble is available in the package data object
gsp4, while the CSV file of it is available at:
system.file("extdata/gsp4.csv", package = "gscramble")Here is what it looks like:
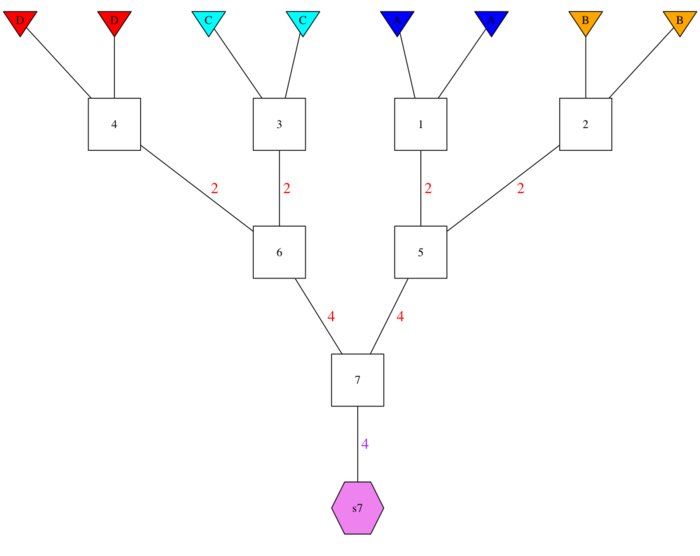
Mapping populations/collections to founding populations.
RepPop
When you create a genomic simulation pedigree, you will typically
denote the groups/populations/clusters that the founders come from with
short names, like “A” or “B”. However, the actual group names used in
your genotype dataset (as specified in I_meta) might be
different. In our example data for this R package, we have 4 groups. To
quickly illustrate how many individuals are included in each of these
example groups:
I_meta %>%
count(group)
#> # A tibble: 4 × 2
#> group n
#> <chr> <int>
#> 1 Pop1 20
#> 2 Pop2 20
#> 3 Pop3 18
#> 4 Pop4 20To define how individuals among groups combine to form hybrids, you
must use a tibble with columns index, pop,
group, to indicate which of the founding populations (“A”,
“B”, etc.) correspond to the different groups (from the
group column in, for example, I_meta) in your
genotype data set; thus, the RepPop serves to
translate/align your I_meta with your GSP.
If you wish to iterate the segregation procedure multiple times in a
single simulation—each time assigning new genetic material to the
founders of the pedigree—you can specify that by doing multiple
replicates of the procedure as defined in the index column.
The index column must be of type integer, and it must
include all values from 1 up to the number of simulation replicates
desired. Rows with the same value,
,
say, of index give the desired mappings from founding pops
(“A”, “B”, etc) in the GSP to different groups in the genotype data for
the
replicates of the simulation.
Each different replicate is made by drawing genetic material
without replacement from the genotypes (Geno in
the example data) and placing that genetic material into the founders of
the GSP. Sampling of alleles from the haplotypes within Geno into the
founders of the GSP is always done without replacement within the
specification of a particular RepPop. (Thus, for example, if you have
indexes from 1 to 20, with each one mapping three A founders to
individuals in Pop1 and four B founders to individuals in Pop2, then you
will need to have genotypes from at least
individuals in Pop1 and
individuals in Pop2).
An example might help. Suppose that we wish to do a simulation with
the pedigree in GSP (13 individuals, 6 of which are
founders: 3 from population “A” and 3 from population “B”). For the
first replicate (index = 1), we might want to map “A” to
Pop1 and “B” to Pop2, and in the second replicate (index =
2) we might want to map “A” to Pop4, and “B” to Pop 3. Note, at this
point, genetic material from 3 individuals from each of those
populations will have been “consumed” from each of these populations and
segregated, without replacement, into the samples from the genomic
simulation pedigree.
The RepPop tibble that would specify this is given in
the package variable RepPop1:
RepPop1
#> # A tibble: 4 × 3
#> index pop group
#> <int> <chr> <chr>
#> 1 1 A Pop1
#> 2 1 B Pop2
#> 3 2 A Pop4
#> 4 2 B Pop3For another example, imagine that we want to do three replicates
(index = 1:3) creating the admixed individuals sampled from
the genomic permutation pedigree, gsp4. The RepPop tibble
for that might look like this:
RepPop4
#> # A tibble: 12 × 3
#> index pop group
#> <int> <chr> <chr>
#> 1 1 A Pop1
#> 2 1 B Pop2
#> 3 1 C Pop3
#> 4 1 D Pop4
#> 5 2 A Pop1
#> 6 2 B Pop2
#> 7 2 C Pop3
#> 8 2 D Pop4
#> 9 3 A Pop1
#> 10 3 B Pop2
#> 11 3 C Pop3
#> 12 3 D Pop4Note that this request will consume one individual from each of populations 1, 2, 3, and 4 (which are mapped to A, B, C, and D, respectively), and will simulate/create 4 admixed individuals.
Segregating Chunks of Genome Without Replacement
Now that we have been through all of the input data formats, we can
use them to segregate chunks of chromosome. Note that this first part
does not require genotypes. We are just simulating the process by which
big chunks of chromosomes drop through the pedigrees. The function used
for this is segregate(). The needed inputs are:
- 1 or more genomic permutation pedigrees (like
GSPorgsp4) - A RepPop tibble to go with each genomic permutation pedigree
- The recombination probabilities, like
RecRates.
The final wrinkle here is that we have set this up so that you can
specify, in a single simulation, that replicates from multiple genomic
permutation pedigrees can be requested. This is done by passing
segregate() a tibble that has a list column named
gpp, which holds the genomic permutation pedigrees, and a
parallel column named reppop, which holds the RepPop
tibbles for each of those genomic permutation pedigrees.
But, let’s start simple, specifying only a reasonably simple genomic
permutation pedigree (gspComplex) and a RepPop tibble with
Pop1 as the focal group (in backcrosses), hybridizing with Pop2. The
tools described below are not that interesting for the most simplistic
GSP (gspF1) in that the simulation of F1s does not provide the
opportunity to illustrate recombination. Accordingly, we are
illustrating segregate() with the slightly more complex
gspComplex, producing F1s, F1BC1s, F2BC1s …
Input_tibble <- tibble(
gpp = list(gspComplex),
reppop = list(RepPopSimpleF1)
)
# here is what that input object looks like:
Input_tibble
#> # A tibble: 1 × 2
#> gpp reppop
#> <list> <list>
#> 1 <tibble [7 × 11]> <tibble [2 × 3]>The segregation requested is then carried out, using the
recombination rates in RecRates like this:
The output from that is a big tibble. Each row represents one segment
of genetic material amongst the sampled individuals from the genomic
permutation pedigrees. Each segment exists in one of the samples
(samp_index) from a sampled individual with a
ped_sample_id on a given gpp (the index giving
the row of the request input tibble) in a given rep within
the individual. Further, it is on one of two gametes
(gamete_index) that segregated into the individual, and it
came from a certain founding population (pop_origin) that
corresponds to the named groups in the genotype file
(group_origin). And, of course, the segment occupies the
space from start to end on a chromosome
chrom. Finally, the index of the founder haplotype on the
given gpp that this segement descended from is given in
rs_founder_haplotype which is short for “rep-specific
founder haplotype”. This final piece of information is crucial for
segregating variation from the individuals in the Geno file
onto these segments. We take that up in the next section, but let’s
first look at the results that we have here.
First, here is a listing of the top of the Segments we
produced above.
Segments
#> # A tibble: 36 × 14
#> chrom_f gpp index chrom ped_sample_id samp_index gamete_index
#> <fct> <int> <int> <chr> <chr> <int> <dbl>
#> 1 12 1 1 12 5 1 1
#> 2 17 1 1 17 5 1 1
#> 3 18 1 1 18 5 1 1
#> 4 12 1 1 12 5 1 2
#> 5 17 1 1 17 5 1 2
#> 6 18 1 1 18 5 1 2
#> 7 12 1 1 12 6 1 1
#> 8 17 1 1 17 6 1 1
#> 9 18 1 1 18 6 1 1
#> 10 12 1 1 12 6 1 2
#> # ℹ 26 more rows
#> # ℹ 7 more variables: gamete_segments <list>, pop_origin <chr>,
#> # rs_founder_haplo <int>, start <dbl>, end <dbl>, group_origin <chr>,
#> # sim_level_founder_haplo <int>The table is a little wide, so we will show the first half of the columns and then the second half.
First half
Segments %>%
select(gpp:pop_origin)
#> # A tibble: 36 × 8
#> gpp index chrom ped_sample_id samp_index gamete_index gamete_segments
#> <int> <int> <chr> <chr> <int> <dbl> <list>
#> 1 1 1 12 5 1 1 <dbl [2]>
#> 2 1 1 17 5 1 1 <dbl [2]>
#> 3 1 1 18 5 1 1 <dbl [2]>
#> 4 1 1 12 5 1 2 <dbl [2]>
#> 5 1 1 17 5 1 2 <dbl [2]>
#> 6 1 1 18 5 1 2 <dbl [2]>
#> 7 1 1 12 6 1 1 <dbl [2]>
#> 8 1 1 17 6 1 1 <dbl [2]>
#> 9 1 1 18 6 1 1 <dbl [2]>
#> 10 1 1 12 6 1 2 <dbl [3]>
#> # ℹ 26 more rows
#> # ℹ 1 more variable: pop_origin <chr>Second half
Segments %>%
select(rs_founder_haplo:group_origin)
#> # A tibble: 36 × 4
#> rs_founder_haplo start end group_origin
#> <int> <dbl> <dbl> <chr>
#> 1 1 0 63582536 Pop1
#> 2 1 0 69302804 Pop1
#> 3 2 0 61201108 Pop1
#> 4 1 0 63582536 Pop2
#> 5 1 0 69302804 Pop2
#> 6 2 0 61201108 Pop2
#> 7 4 0 63582536 Pop1
#> 8 4 0 69302804 Pop1
#> 9 3 0 61201108 Pop1
#> 10 2 0 17363020. Pop1
#> # ℹ 26 more rowsVisualizing those chunks of genome
Let’s glance at the gspComplex (also illustrated above):
For convenience, we built a function called
plot_simulated_chromosome_segments() that let’s you quickly
visualize the results. Let’s try it here. By adding the RecRates to the
function call, we get little sparklines showing us the recombination
rates across the chromosomes.
g <- plot_simulated_chromomsome_segments(Segments, RecRates)
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the gscramble package.
#> Please report the issue at <https://github.com/eriqande/gscramble/issues>.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.
g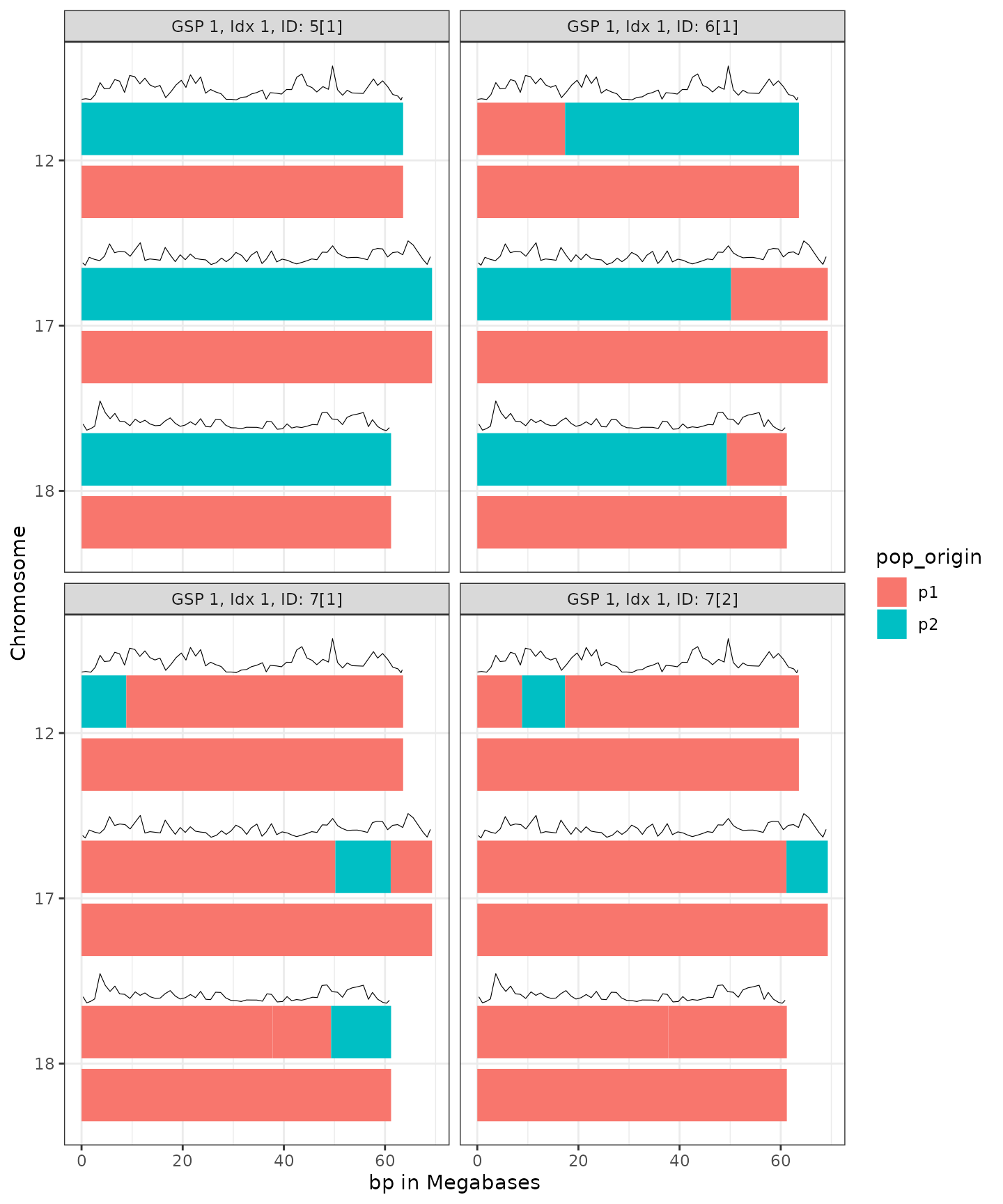
As you can see, Individual #5 (F1 specified in the GSP above)
represents full chromosomes from Pop1 and Pop2, without recombination,
at each of the 3 chromosomes included in the example data. As described
in the GSP, one of the F1s was carried down the pedigree to simulate an
F1BC1 (Individual #6).
The chromosomal composition of Individual 6 illustrates the effect of
recombination.
Similarly, one of the F1BC1s was carried down to simulate an F1BC2
(Individuals #7). Here we have two #7s, illustrating distinct patterns
of recombination.
Simulating alleles at markers within segments
There are a few separate steps required to make this happen, and they
all occur within the function segments2markers(). The steps
that happen are:
- The genotype matrix is reorganized according to the
groupspecification in theI_meta, so that individuals in the same group are adjacent to one another in the genotype matrix. - The alleles carried by the individuals within each population are permuted.
- The chromosomes carrying these permuted alleles from
Genoare mapped to the chromosomes carried by the founders in the simulation performed bysegregate(). - The alleles on the founder chromosomes are propagated to the descendant segments in the individuals sampled from the GSP.
- Missing data is dealt with (if a site has one gene copy missing after the permutation and mapping described above, the other gene copy must be declared missing, as well).
- The output genotypes are formatted for return, along with some information about the true admixture proportions of each individual.
The input to segments2markers() is:
- A tibble of segments, like that returned from
segments(). We could use the variableSegmentsdefined above in this vignette example, but we will re-run thesegregate()step, here, explicitly including the marker meta data. This is helpful because it re-orders chromosomes, if necessary, and checks that all the markers are included within the bounds of the recombination rate bins. - The individual meta data,
- The marker meta data,
- The genotype data.
We invoke it like this:
set.seed(15) # for reproducibility
# re-run segment segregation, explicitly passing it the
# marker meta data
Segments2 <- segregate(
request = Input_tibble,
RR = RecRates,
MM = M_meta
)
Markers <- segments2markers(
Segs = Segments2,
Im = I_meta,
Mm = M_meta,
G = Geno
)The output is a list of three components:
-
Markers$ret_genois an N x 2L matrix of returned genotypes. The two alleles at each locus are in two adjacent columns.dim(Markers$ret_geno) #> [1] 78 200# genotypes of the first 10 individuals at the first 3 markers Markers$ret_geno[1:10, 1:6] #> [,1] [,2] [,3] [,4] [,5] [,6] #> [1,] "G" "G" NA NA "T" "T" #> [2,] "G" "G" NA NA "T" "T" #> [3,] "G" "G" "C" "C" "T" "T" #> [4,] "G" "G" "C" "C" "T" "C" #> [5,] "G" "G" "C" "C" "T" "T" #> [6,] "G" "G" "C" "C" "T" "T" #> [7,] "G" "G" "C" "C" "T" "T" #> [8,] "G" "G" "C" "C" "T" "T" #> [9,] "G" "G" NA NA "T" "T" #> [10,] "G" "G" "C" "C" "T" "T" -
$ret_ids: a tibble with IDs of the individuals corresponding to the rows in theret_genomatrix.# Individual IDs for the first 10 individuals Markers$ret_ids[1:10,] #> # A tibble: 10 × 2 #> group indiv #> <chr> <chr> #> 1 ped_hybs h-1-1-5-1-1 #> 2 ped_hybs h-1-1-6-1-2 #> 3 ped_hybs h-1-1-7-1-3 #> 4 ped_hybs h-1-1-7-2-4 #> 5 Pop1 permed_ID0016971 #> 6 Pop1 permed_ID0016972 #> 7 Pop1 permed_ID0017013 #> 8 Pop1 permed_ID0023170 #> 9 Pop1 permed_ID0035746 #> 10 Pop1 permed_ID0035748There are two columns:
groupandindiv. The first rows include the samples simulated as specified in the GSP. Their group isped_hybs(meaning “sampled hybrids from the pedigree”) and their IDs are in theindivcolumn in the format of anh-followed by a string with replaced values of:gpp-rep-ped_sample_id-samp_index-matrix_row. Let’s break down the nomenclature we used to label simulated gentype. Recall with this simulation, we specified only a singleGSP(in the Input_tibble gpp = list(gspComplex).
With thisgspComplex, we are using 3 founders from Pop1 and 1 founder from Pop2 to simulate one F1 (Individual #5 as described withgspComplexabove), one F1BC1 (Individual #6) and two F1BC2s (Individual #7). Within the illustration ofgspComplex, as called from the Input_tibble…Those are:
-
index: theindexvalue from theRepPopobject (only a singleindexin this example) -
ped: the number corresponding to the unique GSP used in the simulation (recall that you can specify multiple GSPs by passingsegregate()a list namedgppwhich holds the genomic permutation pedigrees used in the simulation, but we only passed 1 GSP in that list), -
sample_id: corresponding sample number from the GSP (recall that multiple individuals simulated within the pedigree [non-founders] may share the same sample number, but these IDs correspond to #5, #6, and two #7s withgspComplex) -
samp_index: indexing of individuals withinsample_ids (one #5[F1], one #6 [F1BC1], and two #7s [F1BC2]) -
matrix_row: the row number of the genotype matrix
After the individual hybrid samples from the pedigree, there are rows of all the remaining individuals whose genotypes after permutation were not involved as founders in the segregation simulation. These are given the name
permed+ their original IDs. The group that each belongs to is in the group column. Things are organized this way so that the whole genotype can easily be passed to ADMIXTURE and the admixture fractions of the hybrid individuals estimated using thepermed_*individuals as samples of known cluster/group origin. -
$hyb_Qs: A tibble with the true admixture fraction (calculated as the total proportion of genome length from each group) of each simulated hybrid.
Now, briefly, we can illustrate this again with more complexity.
Let’s illustrate the capacity to simulate multiple genomic permutation
pedigrees (GSP and gsp4 illustrated below) by passing
segregate() a tibble that has a list column named
gpp, which holds the genomic permutation pedigrees, and a
parallel column named reppop, which holds the RepPop
tibbles for each of those genomic permutation pedigrees.
Input_tibble <- tibble(
gpp = list(GSP, gsp4),
reppop = list(RepPop1, RepPop4)
)
# here is what that input object looks like:
Input_tibble
#> # A tibble: 2 × 2
#> gpp reppop
#> <list> <list>
#> 1 <tibble [13 × 11]> <tibble [4 × 3]>
#> 2 <tibble [7 × 11]> <tibble [12 × 3]>
g <- plot_simulated_chromomsome_segments(Segments, RecRates)
g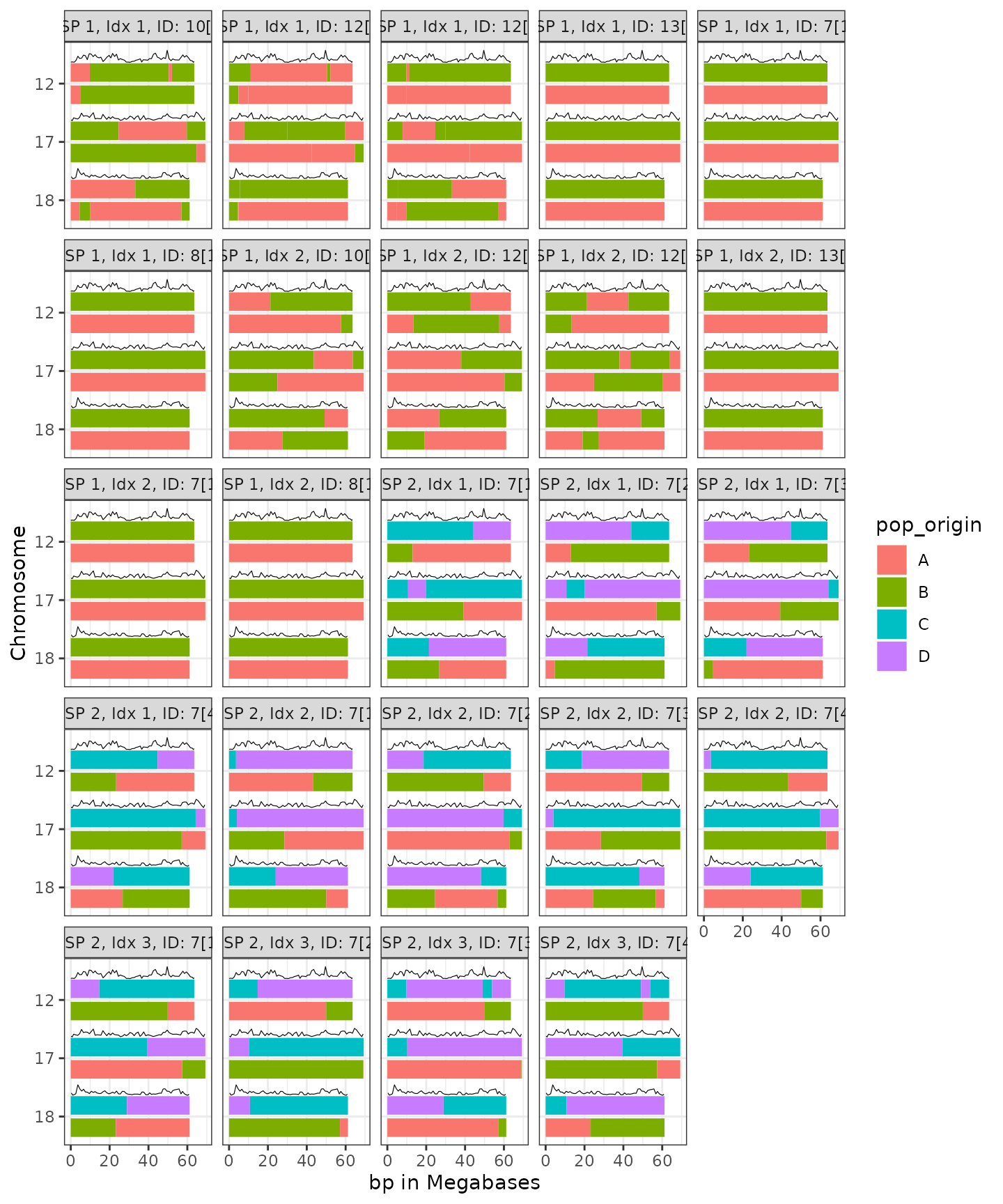
Markers <- segments2markers(
Segs = Segments,
Im = I_meta,
Mm = M_meta,
G = Geno
)To and From plink Format
Now that we have illustrated the application of ‘gscramble’ with this
simple dataset and the included R objects Geno,
I_meta, and M_meta, you are probably eager to
try ‘gscramble’ with data of your own. Use the function
plink2gscramble() to directly input genotypes in the PLINK
(.ped / .map) format. You must pass the function
plink2gscramble() both the .ped and corresponding .map
files as illustrated below. If your data is in a binary PLINK format
(.bed / .bim / .fam), first use PLINK (option --recode) to
export .ped and .map files that are accessible for ’gscramble’s
plink2gscramble().
####### These lines are not portable, since gscrambleTutorial.ped and
####### gscrambleTutorial.map are not available
# with .ped / .map files
#plinkIN <- plink2gscramble(ped = "gscrambleTutorial.ped", map = "gscrambleTutorial.map")
#ls(plinkIN)
#str(plinkIN)
#dim(plinkIN$Geno)
#plinkIN$Geno[1:3,1:10]
#rm(plinkIN)
# identifical result with corresponding .ped.gz / .map.gz files
###### End non-portable lines
map_plink <- system.file("extdata/example-plink.map.gz", package = "gscramble")
ped_plink <- system.file("extdata/example-plink.ped.gz", package = "gscramble")
plinkIN <- plink2gscramble(ped_plink, map_plink)
dim(plinkIN$Geno)
#> [1] 78 200
dim(plinkIN$I_meta)
#> [1] 78 6
dim(plinkIN$M_meta)
#> [1] 100 4Or, if you want to just pass the prefix to the ped and the map files, you could use:
prefix <- system.file("extdata/example-plink.map.gz", package = "gscramble")
prefix <- str_replace(prefix, "\\.map\\.gz", "")
plinkPRE <- plink2gscramble(prefix = prefix, gz_ext = TRUE)
dim(plinkPRE$Geno)
#> [1] 78 200
dim(plinkPRE$I_meta)
#> [1] 78 6
dim(plinkPRE$M_meta)
#> [1] 100 4Here, we use the gz_ext = TRUE option because our files
are gzipped with the .gz extension.
When it is time to write out a set of plink files, you can use
gscramble2plink():
# note, we are only using a temporary file here for the output
# because this is in the vignette of an R package. You, yourself,
# will likely want to write the results to a directory in your
# home directory somewhere, like `prefix = ~/my_stuff/gscram-sim-1`, etc.
tfile <- tempfile()
gscramble2plink(
I_meta = Markers$ret_ids,
M_meta = M_meta,
Geno = Markers$ret_geno,
prefix = tfile
)
#> pedfile written to /tmp/Rtmp2djsu4/file1e3030c34c6d.ped
#> mapfile written to /tmp/Rtmp2djsu4/file1e3030c34c6d.map
#> [1] TRUE